

Abstract
Autism spectrum disorder (ASD) comprises a group of neurodevelopmental disorders characterized by social deficits and stereotyped behaviors. While several theories have emerged, the pathogenesis of ASD remains unknown. Although studies report dopamine signaling abnormalities in autistic patients, a coherent dopamine hypothesis which could link neurobiology to behavior in ASD is currently lacking. In this paper, we present such a hypothesis by proposing that autistic behavior arises from dysfunctions in the midbrain dopaminergic system. We hypothesize that a dysfunction of the mesocorticolimbic circuit leads to social deficits, while a dysfunction of the nigrostriatal circuit leads to stereotyped behaviors. Furthermore, we discuss 2 key predictions of our hypothesis, with emphasis on clinical and therapeutic aspects. First, we argue that dopaminergic dysfunctions in the same circuits should associate with autistic-like behavior in nonautistic subjects. Concerning this, we discuss the case of PANDAS (pediatric autoimmune neuropsychiatric disorder associated with streptococcal infections) which displays behaviors similar to those of ASD, presumed to arise from dopaminergic dysfunctions. Second, we argue that providing dopamine modulators to autistic subjects should lead to a behavioral improvement. Regarding this, we present clinical studies of dopamine antagonists which seem to have improving effects on autistic behavior. Furthermore, we explore the means of testing our hypothesis by using neuroreceptor imaging, which could provide comprehensive evidence for dopamine signaling dysfunctions in autistic subjects. Lastly, we discuss the limitations of our hypothesis. Along these lines, we aim to provide a dopaminergic model of ASD which might lead to a better understanding of the ASD pathogenesis.
Introduction
Autism spectrum disorder (ASD) comprises a heterogeneous group of neurodevelopmental disorders, the core features of which are persistent deficits in social interaction and communication and restricted patterns of behavior, interests, and activities [1]. Although various theories have been proposed, the pathogenesis of ASD remains largely unknown [2]. This is due to the spectrum's extreme heterogeneity, spanning from social to nonsocial behavioral anomalies.
Similar to other neuropsychiatric disorders with shared behavioral traits, such as schizophrenia [3], evidence suggests that ASD might be linked to dopaminergic dysfunctions [4,5,6]. Indeed, numerous authors have put forth the idea that dopamine imbalances in specific brain regions could lead to autistic-like behavior, as reviewed in great detail by Dichter et al. [7]. However, existing views fail to clarify the role of dopamine signaling anomalies in prompting the behavioral features of ASD. Thus, despite extensive research into the matter, we must admit that a dopamine hypothesis of ASD is currently lacking.
Given these facts, we further propose a framework for such a hypothesis. First, we argue how specific dopaminergic dysfunctions could lead to autistic-like behavior. Second, we provide a comprehensive discussion of 2 key predictions of our hypothesis. Third, we indicate how our hypothesis might be tested. Lastly, we discuss the limitations of the hypothesis. In this way, we aim to provide a clear dopaminergic model of ASD.
How Could Dopaminergic Dysfunctions Lead to Autistic-Like Behavior?
As previously stated, autistic subjects display social deficits and stereotyped behaviors as core traits. We propose that these traits arise from altered midbrain dopaminergic signaling. Two subpopulations of midbrain dopaminergic neurons are involved in controlling the functions traditionally affected in ASD: the ventral tegmental area and the substantia nigra [8]. These 2 groups of neurons are the origin of 2 diffuse modulatory systems which form broadly scattered connections throughout the brain. Thus, they modulate vast assemblies of postsynaptic neurons [9] and are capable of influencing key behavioral aspects.
First, neurons from the ventral tegmental area project to the prefrontal cortex and the ventral striatum's nucleus accumbens, forming the mesocorticolimbic (MCL) circuit, which is involved in high-order brain functions such as reward and motivation-related behavior [10,11]. Second, neurons from the substantia nigra project towards the dorsal striatum, forming the nigrostriatal (NS) circuit, which controls the motor aspects of goal-directed behavior in order to develop appropriate actions towards obtaining a specific outcome [8,11]. Considering these, we further suggest that autistic core traits arise from the alteration of dopamine signaling in these brain areas.
To begin with, the social deficits observed in ASD could reflect an MCL circuit dysfunction. Given its role in reward and motivation [9], an MCL circuit dysfunction could lead to altered reward representation and reduced motivation to pursue rewarding experiences. If these alterations concerned social behavior, autistic brains might fail to register social experiences as rewarding, further reducing the motivation to seek social interactions and develop social abilities. This view is articulated by the social motivation theory [11] of ASD which states that autistic subjects are an extreme case of reduced social motivation which impacts social cognition, ultimately leading to social deficits.
Several studies provide support for this view. At first, autistic subjects display signaling alterations in the MCL dopaminergic pathway, such as reduced release of dopamine in the prefrontal cortex and reduced neural response in the nucleus accumbens [4,5]. Accordingly, studies show that ASD is characterized by general hypoactivation of the reward system [6], which occurs for both social and nonsocial rewards [4,12]. Reduced mesolimbic activation was also observed in subjects carrying a polymorphism of the ASD-associated oxytocin receptor gene [13], suggesting a facilitating role of oxytocin in the MCL dopaminergic signaling. The improvement of social deficits secondary to intranasal oxytocin administration [14] suggests a determining role of mesolimbic dopamine signaling anomalies in generating the social features of ASD. Lastly, impaired mesolimbic dopaminergic signaling was shown to alter specific reward-related behavior in autistic subjects, such as effort-based decision making for rewards [15]. Altogether, these studies suggest that autistic subjects display general dysfunction of the MCL circuit and subsequent altered reward-related behavior. Therefore, these could represent the initial events of a pathologic cascade, leading to the social deficits observed in ASD.
Next, the stereotyped behaviors observed in autistic patients could arise from a dysfunction of the NS pathway, which has been shown to be involved in mediating stereotypies [16]. Considering its central role in controlling goal-directed motor behavior [9], an NS pathway dysfunction could lead to autistic-like behavior by entrapment into loops of purposeless, stereotyped patterns of behavior.
Evidence to support this view comes mainly from mouse model studies. Given that drug-induced NS pathway dysfunction led to stereotypies in mice, Lewis et al. [17] postulate that stereotypic behavior is a result of a dysfunctional NS circuit. Furthermore, administration of intrastriatal D1 dopamine receptor antagonists led to attenuation of spontaneous stereotypies in mice [18], pointing to a critical role of dopamine signaling anomalies in mediating stereotypic behavior. Furthermore, Horev et al. [19] studied a mouse model for 16p11.2 deletion, a mutation found in patients with ASD. Mice carrying the mutation displayed NS lesions associated with stereotyped patterns of behavior, generating the so-called behavior trap phenotype.
In autistic subjects, a polymorphism of the D3 dopamine receptor (DR3) gene was associated with a greater striatal volume and severity of stereotypic behavior [20,21]. Moreover, common polymorphisms of the dopamine 4 receptor (DR4) gene and the dopamine transporter (DAT) gene were associated with repetitive behavior in children with ASD [22]. Altogether, these studies suggest a determining role for the dopaminergic dysfunction of the NS pathway in eliciting autistic-like stereotypic behavior.
In this dopamine hypothesis framework, dopamine signaling abnormalities in the diffuse dopaminergic modulatory systems of the midbrain lead to social deficits and stereotyped behaviors (Fig. 1), providing a link between neurobiology and behavior in ASD.
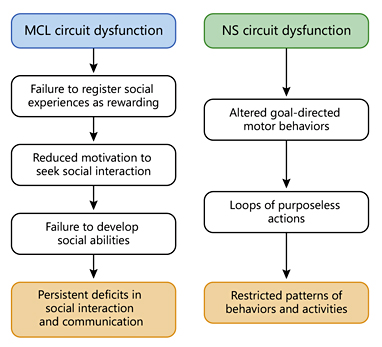
The proposed mechanism through which dysfunctions of the diffuse dopaminergic modulatory systems of the midbrain could lead to autistic core features.
The proposed mechanism through which dysfunctions of the diffuse dopaminergic modulatory systems of the midbrain could lead to autistic core features.
This view yields 2 essential predictions. First, dopaminergic dysfunctions in the same brain areas should associate with autistic-like behavior regardless of the diagnosis (i.e., in nonautistic subjects). Second, supplying dopamine modulators to autistic subjects should lead to behavioral improvement. Below, we further provide a discussion over these predictions of our hypothesis. In relation to our first prediction, we present the case of PANDAS (pediatric autoimmune neuropsychiatric disorder associated with streptococcal infections).
PANDAS: A Clinical Bridge between Dopamine and ASD
PANDAS is a neuropsychiatric disorder which has been proposed as a variant of Sydenham chorea, a widely recognized poststreptococcal autoimmune disorder [23]. It is thought that both disorders have a common pathogenic mechanism, although PANDAS has predominantly psychiatric symptoms [24]. Affected subjects display social deficits and restricted patterns of behavior, traits which are strikingly similar to those exhibited by autistic patients [1].
The pathogenesis of PANDAS involves molecular mimicry: host antibodies which cross-react with endogenous proteins in the brain, specifically found in the striatum [24]. Autoantibodies from patients with PANDAS were shown to bind to dopaminergic neurons and cause functional alterations. Evidence shows that antibody binding alters the sensitivity of receptors [25], suggesting a role of dopamine signaling anomalies in prompting the behavioral traits in PANDAS. Furthermore, studies show that PANDAS antibodies activate the Ca2+/calmodulin-dependent protein kinase II (CaMKII) signaling pathway [26]. CaMKII is a protein kinase involved in various signaling cascades, with key roles in high-order brain functions [27]. In dopaminergic neurons, CaMKII binds directly to D3 dopamine receptor (D3R) and downregulates its function. Given that D3R is preferentially expressed in the striatum, antibody binding could alter reward, motivation, and motor-related behavior in PANDAS patients [28]. Intriguingly, several studies support the role of D3R and CaMKII signaling dysfunctions in ASD [20,21,29,30], indicating a shared pathogenesis between PANDAS and ASD.
Altogether, these data suggest that altering dopamine neurotransmission in the striatum might lead to behavioral traits strikingly similar to those of ASD [24]. This provides support for the first prediction yielded by our hypothesis: dopaminergic dysfunctions in the same brain areas associate with autistic-like behavior regardless of diagnosis. Next, we look at the second prediction of our hypothesis, regarding the behavioral improvement in autistic subjects treated with dopamine antagonists.
Dopamine Antagonists: A Pharmacological Bridge between Dopamine and ASD
As previously stated, some authors proposed models regarding dopamine imbalances in autistic subjects, concerning either the hyperactivity or hypoactivity of dopaminergic pathways [31,32]. Regardless of these models, no consensus has been reached. Schizophrenia, which shares behavioral traits with ASD [3], is thought to arise from the joint action of subcortical dopamine excess and cortical dopamine deficit [33]. Given the shared behavioral traits between schizophrenia and ASD, a somewhat similar pathogenic mechanism is likely to occur in autistic patients as well. Unfortunately, evidence is currently limited and further studies are necessary in order to set up a coherent dopaminergic model of ASD, which would further lay the foundation for an adequate treatment.
Indeed, the importance of a dopamine hypothesis of ASD resides in the prospect of treatment with dopamine antagonists. Among these, risperidone and aripiprazole are the only agents approved by the Food and Drug Administration for the management of ASD-associated behavior [34]. Even though these drugs are prescribed mainly for treating irritability in autistic patients, there have been several studies that assessed their effects on the core traits of ASD. Thus, risperidone was shown to have a significant effect on stereotyped behavior in autistic subjects [35,36], although not seen in all studies [37]. Intriguingly, risperidone also improved social behavior, as evaluated by behavioral scales such as the Childhood Autism Rating Scale [37,38]. However, these results must be regarded with caution, since the extreme heterogeneity of the disorder limits the statistical power of outcome measures in clinical trials [39].
The observed therapeutic effect of dopamine antagonists on the autistic core traits is essential in providing support for the second prediction of our hypothesis: if autistic-like behavior arises from dopaminergic dysfunctions, then dopamine modulators should lead to both social and nonsocial behavioral improvement. Blocking dopamine neurotransmission could also prove to be an important therapeutic test, since it leads to behavioral improvement. However, studies are inconsistent and suggest that autistic subjects display general dopaminergic hypoactivity [4,5,6]. Yet, evidence is scarce and contradictory data pose the need for additional studies in order to establish the characteristics of dopamine signaling in ASD. Along these lines, we will further explore ways of testing the dopamine hypothesis of ASD.
Testing the Dopamine Hypothesis of ASD
Our dopamine hypothesis of ASD is falsifiable and can be rigorously tested by using neuroreceptor imaging. This method is employed through imaging techniques, such as SPECT (single photon emission computerized tomography) and PET (positron emission tomography), and involves injecting radioactively labelled agents (radiotracers) that will bind to specific neuroreceptors [40]. Using neuroreceptor imaging in autistic subjects could provide several lines of evidence for dopamine dysfunctions [33], as we shall argue further. Ideally, studies should compare untreated autistic subjects, dopamine antagonist-treated autistic subjects, and neurotypical controls. Only a few neuroreceptor imaging studies have investigated dopamine in ASD, and no consensus across studies has been reached [41].
First, by using competition between endogenous dopamine and radiotracers, neuroreceptor imaging studies should examine dopamine levels at baseline and after amphetamine challenge, which produces dopamine release into the synapses [42]. Thus, results could indicate whether autistic subjects display an altered baseline dopamine level or altered release of dopamine in response to stimuli and whether treatment with dopamine antagonists has any effect over these alterations. Moreover, amphetamine challenge would be an important opportunity to note whether autistic core symptoms modify in relation to synaptic dopamine release.
Second, studies should assess the density and occupancy (hence availability) of dopamine receptors in both subcortical and cortical areas. Results from these studies could show whether autistic subjects display differences in D2R and/or D1R availability. Divergent availability of dopamine receptors could indicate a contrasting subcortical/cortical response to dopamine, as already hypothesized to occur in schizophrenia [33]. Moreover, these studies could indicate whether dopamine antagonists significantly alter the autistic brains' responsivity to dopamine.
Altogether, these studies could provide comprehensive evidence for dopamine signaling dysfunctions and the effects of dopamine antagonists on these alterations. These data could set up a clear dopaminergic model of ASD, which would further provide effective means of treatment. In the following section, we discuss the limitations of our hypothesis.
Limitations of the Dopamine Hypothesis of ASD
First, we proposed a pathophysiological approach through which we linked dopamine dysfunctions in essential circuits to autistic-like behavior. We did not intend to provide an etiologic framework, as ASD is currently considered a disorder with multiple etiologies that converge to alterations in brain circuits [43]. Thus, it is likely that diverse molecular syndromes assemble in different amounts and induce signaling anomalies in specific brain circuits, such as those proposed by our hypothesis.
Second, our hypothesis aims to provide a model for the autistic core features, not for the entire variation of the spectrum, which includes numerous comorbidities [44]. It is likely that other neurobiological anomalies co-occur, given the spectrum's extreme heterogeneity. Nonetheless, dopamine signaling anomalies have been associated with several ASD comorbidities, such as ADHD, anxiety, tics [45], and executive function deficits [46].
Lastly, we do not intend to present ASD as a dopamine signaling disorder. It is likely that other neurotransmitter signaling anomalies contribute to the neurobiological underpinnings of ASD. As a matter of fact, a recent paper hypothesizes an important role of aberrant glutamate signaling in ASD pathophysiology [47]. Thus, it is likely that autistic-like behavior arises from dysfunctions in specific brain circuits, set in motion by diverse neurotransmitter signaling anomalies.
Conclusions
Although several dopamine dysfunctions have been reported in autistic patients, a dopamine hypothesis of ASD per se is currently lacking. We have presented such a hypothesis by proposing that autistic-like behavior arises from dopamine dysfunctions in the diffuse dopaminergic modulatory systems of the midbrain, with an impact on social motivation and goal-directed motor behavior. In this way, we provide a link between neurobiology and behavior in ASD.
This hypothesis framework provides 2 essential predictions. First, dopamine dysfunctions in specific brain areas should lead to social deficits and stereotyped behaviors, regardless of diagnosis (i.e., in nonautistic subjects). Concerning this, we considered the case of PANDAS, a neuropsychiatric disorder with behavioral features strikingly similar to those of ASD. The behavioral features of PANDAS are thought to arise from alterations of dopamine signaling in circuits hypothesized as defective by our hypothesis. By confirming this essential prediction, PANDAS generates a clinical bridge towards a dopamine hypothesis of ASD. Second, if autistic-like behavior is due to dopamine dysfunctions, then dopamine modulators should lead to behavioral improvement. The fact that dopamine antagonists have proved to be efficient in improving autistic core behavior confirms this prediction and generates a pharmacological bridge between dopamine and ASD. Altogether, these data provide support for our dopaminergic model of ASD.
Moreover, our hypothesis can be thoroughly tested by using neuroreceptor imaging studies, which could provide information about synaptic dopamine levels and the availability of dopamine receptors in both subcortical and cortical structures. Overall, these studies could provide comprehensive evidence for the hypothesized dopamine signaling alterations in autistic subjects.
Even though our dopamine hypothesis of ASD has limitations, it could open perspectives for generating a clear pathogenetic model of ASD. In turn, this model might provide means for an effective treatment with dopamine antagonists, which seem to have improving effects on autistic-like behavior. Moreover, uncovering dopaminergic dysfunctions in autistic patients could offer the possibility to extend knowledge in disorders with shared behavioral traits, such as schizophrenia and PANDAS. Finally, studying the pathogenesis of neuropsychiatric disorders might lead to a deeper understanding of the biological basis of human behavior.
Acknowledgements
The author would like to thank Nicoleta Gherghel, MD and Andrei Hopulele-Petri, MD (“Iuliu Hațieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania) for their support and insightful comments on this work.
