

Abstract
Fragile X syndrome (FXS) is a neurodevelopmental disorder characterized by intellectual disability, sensory hypersensitivity, and high incidences of autism spectrum disorders and epilepsy. These phenotypes are suggestive of defects in neural circuit development and imbalances in excitatory glutamatergic and inhibitory GABAergic neurotransmission. While alterations in excitatory synapse function and plasticity are well-established in Fmr1 knockout (KO) mouse models of FXS, a number of recent electrophysiological and molecular studies now identify prominent defects in inhibitory GABAergic transmission in behaviorally relevant forebrain regions such as the amygdala, cortex, and hippocampus. In this review, we summarize evidence for GABAergic system dysfunction in FXS patients and Fmr1 KO mouse models alike. We then discuss some of the known developmental roles of GABAergic signaling, as well as the development and refinement of GABAergic synapses as a framework for understanding potential causes of mature circuit dysfunction. Finally, we highlight the GABAergic system as a relevant target for the treatment of FXS.
Introduction
Fragile X syndrome (FXS) is the most common form of inherited intellectual disability and a leading genetic cause of autism [de Vries et al., 1998; Hagerman et al., 2009]. The genetic basis of FXS is a CGG repeat expansion mutation in the Fmr1 gene [Verkerk et al., 1991], which codes for fragile X mental retardation protein (FMRP), an RNA-binding protein involved in the transport and translational regulation of a wide array of cellular and synaptic proteins [Bagni and Greenough, 2005; Bassell and Warren, 2008]. In addition to cognitive disability and features of autism spectrum disorders (ASD), FXS patients are characterized by anxiety, sensory hypersensitivity, and elevated incidences of epilepsy [Berry-Kravis, 2002; Berry-Kravis et al., 2010; Hagerman and Stafstrom, 2009; Hagerman et al., 2009; Miller et al., 1999; Musumeci et al., 1999], consistent with circuit dysfunction and imbalances in excitatory and inhibitory transmission in various brain regions, including the amygdala, cortex, and hippocampus. Excitatory-inhibitory imbalances and synaptic dysfunction are proposed to play a central role in a number of pathological states, including FXS and ASD [Belmonte and Bourgeron, 2006; Moy and Nadler, 2008; Rubenstein and Merzenich, 2003; Zoghbi, 2003].
Inhibitory neurotransmission in the CNS is mediated by populations of neurons that synthesize and release GABA, which exerts its effects through two subtypes of receptor: the ionotropic GABAA receptors and the metabotropic G-protein-coupled GABAB receptors. In mature neurons, GABAA receptors mediate postsynaptic membrane hyperpolarization by permitting intracellular Cl– influx in the presence of GABA. These postsynaptic receptors comprise two categories: synaptic receptors that produce fast, phasic inhibition in response to relatively high concentrations (mM) of synaptically released neurotransmitter, and extrasynaptic receptors that produce a slow, persistent tonic conductance in response to low neurotransmitter concentrations (nM to low µM) in the extrasynaptic space [for a review, see Farrant and Nusser, 2005]. GABAB receptor activation likewise hyperpolarizes the postsynaptic membrane by activating G-protein-coupled inwardly rectifying K+ channels to produce slow inhibitory currents [Padgett and Slesinger, 2010]. In addition to its role as a postsynaptic inhibitory neurotransmitter, GABA can also modulate neurotransmitter release in an autocrine or paracrine fashion, via distinct mechanisms at presynaptic GABAA and GABAB receptors [Bettler et al., 2004; Trigo et al., 2008]. GABAergic neurons exhibit striking morphological, biochemical, and physiological diversity [Kawaguchi and Kubota, 1997; Markram et al., 2004; Somogyi and Klausberger, 2005]. These populations are further defined by their tendency to form distinct inhibitory circuits based on reciprocal electrical connectivity [Beierlein et al., 2003; Galarreta and Hestrin, 1999; Gibson et al., 1999], as well as their propensity to synapse onto a specific subcellular compartment of target neurons [Kawaguchi and Kubota, 1997; Muller et al., 2006, 2007; Somogyi et al., 1998], indicating a high degree of circuit refinement in the mature brain. This refinement is further indicated by the fact that inhibitory synapses at different subcellular compartments are defined by the expression of different GABAA receptor subunits [Fritschy and Brunig, 2003; Fritschy et al., 1998; Klausberger et al., 2002; Nusser et al., 1996; Nyiri et al., 2001]. Thus, GABAergic inhibitory neurons comprise highly specific circuits acting to control cellular and network excitability, direct the flow of information, and generate neural synchrony and oscillations involved in a number of cognitive processes [Cardin et al., 2009; Porter et al., 2001; Sohal et al., 2009; Wang, 2010]. Given these crucial roles of inhibitory neurons in normal circuit function, it is not surprising that interneuron loss or dysfunction has been implicated in a number of disease states, including FXS, ASD, Rett syndrome, schizophrenia, and epilepsy [Buckmaster and Jongen-Rêlo, 1999; Chao et al., 2010; Cossart et al., 2001; Gogolla et al., 2009; Lewis et al., 2005; Selby et al., 2007].
In view of the importance of inhibitory transmission in normal circuit function and its relevance to a number of neurodevelopmental disorders, we discuss elements of the GABAergic system that are disrupted in the Fmr1 knockout (KO) mouse model of FXS, with a particular focus on postsynaptic GABAA receptor-mediated transmission, emphasizing their relevance for the neurological phenotypes of the syndrome. In addition, given the importance of GABA in synapse and circuit development and maturation [Akerman and Cline, 2007; Ben-Ari, 2002; Huang, 2009], we discuss how defects in this system might contribute to abnormal circuit development. Finally, we summarize studies in patients and animal models that illuminate the GABAergic system as a viable and important therapeutic target for the treatment of FXS.
Crucial GABAergic Synapse Components Are Dysregulated in FXS
FMRP is widely expressed throughout the brain, in both neurons and glia [Devys et al., 1993; Feng et al., 1997; Olmos-Serrano et al., 2010; Wang et al., 2004], and deletion of FMRP results in widespread alterations in the expression of mRNA and proteins from a number of functional categories, including cation channels, adhesion molecules, neurotransmitter receptors, and components of the vesicular transport and release machinery [Brown et al., 2001; Liao et al., 2008; Schütt et al., 2009], suggesting broad cellular and synaptic alterations in the FXS brain. Importantly, FMRP is broadly expressed in GABAergic neuron populations [Feng et al., 1997; Olmos-Serrano et al., 2010], indicating that it is involved in normal interneuron maturation and function.
The GABAergic system was first implicated in the pathogenesis of FXS based on studies of GABAA receptor expression in Fmr1 KO mice. Functional GABAA receptors are heteropentamers, and subunit composition is an important determinant of inhibitory transmission, dictating receptor characteristics such as response kinetics, subcellular localization, and sensitivity to a number of clinically important compounds [Hevers and Lüddens, 1998; Rudolph and Möhler, 2006; Sieghart and Sperk, 2002]. Following the initial finding that FMRP can bind δ subunit mRNA [Miyashiro et al., 2003], a number of studies of mRNA expression in the brains of Fmr1 KO mice revealed prominent reductions in the expression of δ, as well as α, β, and γ subunits in behaviorally relevant brain regions such as cortex and hippocampus [D’Hulst et al., 2006; Gantois et al., 2006]. Complementing these changes in receptor mRNA expression, decreases in α1, α5, β, and δ subunit protein expression [Adusei et al., 2010; Curia et al., 2008; El Idrissi et al., 2005] have been reported, as well as a reduction in gephyrin mRNA [D’Hulst et al., 2009], providing molecular evidence for alterations in inhibitory postsynaptic function in the FXS brain. On the presynaptic side, where FMRP is also expressed [Christie et al., 2009], both increases and decreases in the expression of the rate-limiting GABA-synthesizing enzyme GAD have been reported in Fmr1 KOs [Adusei et al., 2010; D’Hulst et al., 2009; El Idrissi et al., 2005; Olmos-Serrano et al., 2010], with the nature of the change relying, at least in part, on the brain region examined (for example, Olmos-Serrano et al. [2010] report a pronounced decrease in GAD65/67 protein expression in the basolateral amygdala, complemented by decreased vesicular GABA levels, whereas El Idrissi et al. [2005] describe increased GAD65/67 in the cortex, hippocampus, brainstem, and diencephalon, in conjunction with reduced β subunit expression). In addition, proteins required for GABA transport (GAT) and catabolism (GABA-T, SSADH) also exhibit decreased expression in a number of regions [Adusei et al., 2010; D’Hulst et al., 2009; Liao et al., 2008].
Taken together, the general picture provided by these studies is one of extensive dampening of the GABAergic system in FXS, wherein prominent changes in postsynaptic GABAA receptor expression act in combination with alterations in GABA production, metabolism, and release to significantly modify GABAergic function in FXS (fig. 1; table 1).
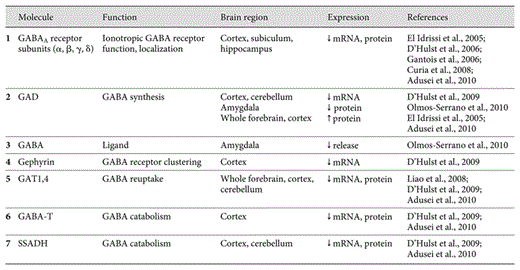
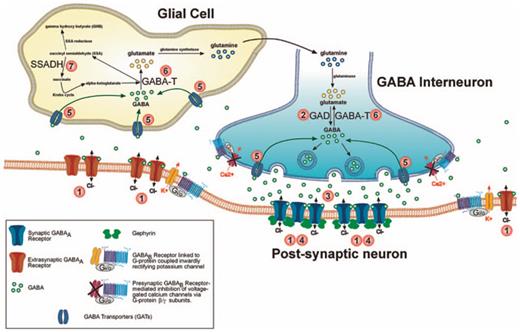
Several GABAergic synapse components exhibit altered expression in the Fmr1 KO mouse model of FXS. Numbers identify key synaptic proteins disrupted in Fmr1 KOs, including GABAA receptors, enzymes involved in GABA production and catabolism (GAD, SSADH, GABA-T) and GAT (inset legend; table 1).
Several GABAergic synapse components exhibit altered expression in the Fmr1 KO mouse model of FXS. Numbers identify key synaptic proteins disrupted in Fmr1 KOs, including GABAA receptors, enzymes involved in GABA production and catabolism (GAD, SSADH, GABA-T) and GAT (inset legend; table 1).
Inhibitory Deficits in Fmr1 KO Mice Are Pervasive, but Region-Specific
Alongside extensive changes in the expression of crucial components of the GABAergic system, Fmr1 KO mice exhibit broad, variable, and distinct disruptions in functional inhibitory transmission in a number of behaviorally relevant brain regions. These observed alterations are highly region-specific, in accordance with the unique function and circuit properties of these individual structures (fig. 2).
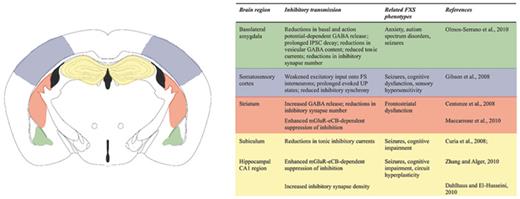
Alterations in inhibitory neurotransmission in the Fmr1 KO mouse brain are pervasive, but region-specific. Regional inhibitory deficits are associated with a number of common FXS phenotypes (inset table). Affected regions include the basolateral amygdala, cerebral cortex, striatum and hippocampus/subiculum. Color coding in the illustration corresponds to the adjacent inset table.
Alterations in inhibitory neurotransmission in the Fmr1 KO mouse brain are pervasive, but region-specific. Regional inhibitory deficits are associated with a number of common FXS phenotypes (inset table). Affected regions include the basolateral amygdala, cerebral cortex, striatum and hippocampus/subiculum. Color coding in the illustration corresponds to the adjacent inset table.
One region of the brain of particular relevance for the behavioral phenotype of FXS is the amygdala, a limbic structure involved in the processing of emotionally salient information and in the acquisition, storage, and extinction of fear memories [Ehrlich et al., 2009; Ledoux, 2003]. The processing of this complex information within this structure relies heavily on inhibitory circuit function [Ehrlich et al., 2009]. In line with evidence of amygdala dysfunction in FXS patients and mouse models alike [Budimirovic et al., 2006; Hagerman et al., 2009; McNaughton et al., 2008; Paradee et al., 1999], the basolateral amygdala of Fmr1 KO mice is characterized by broad and severe inhibitory defects, including reductions in the frequency and amplitude of spontaneous and miniature inhibitory postsynaptic currents (IPSCs), changes in IPSC kinetics, decreased GAD expression and vesicular GABA, and impaired GABA release [Olmos-Serrano et al., 2010]. These alterations in synaptic function are accompanied by a reduction in inhibitory synapse number, indicating aberrant circuit development. In addition to severe deficits in fast synaptic phasic inhibition, Fmr1 KOs also exhibit a profound reduction in tonic inhibition, a major form of GABAergic inhibition in the CNS that relies on GABA availability (determined in part by GABA synthesis, release, uptake, and metabolism), as well as the expression of perisynaptic and extrasynaptic GABAA receptors containing subunits with a high affinity for GABA, such as δ and α5 [Farrant and Nusser, 2005; Semyanov et al., 2004].
The strong, persistent inhibitory conductance provided by tonic inhibition acts to reduce membrane excitability, and thus is an important determinant of cellular output [Semyanov et al., 2004], as illustrated by the occurrence of hippocampal CA3 region hyperexcitability in GABAA receptor α5 subunit KO mice, for example [Glykys and Mody, 2006]. Given the evidence for altered GABA levels and reduced expression of δ and α5 GABAA receptor subunits in Fmr1 KOs, tonic inhibition is expected to be broadly compromised in the FXS brain. In agreement with the findings in the amygdala, recordings from subicular neurons in Fmr1 KOs also reveal a prominent decrease in tonic inhibition, concurrent with regional reduction in δ and α5 mRNA and protein expression [Curia et al., 2008]. In contrast to the amygdala, however, synaptic inhibition is unaltered, indicating circuit-specific effects of FMRP deletion. Broadly speaking, these studies in the amygdala and the subiculum reveal that dampened inhibitory transmission may indeed be a hallmark of the FXS brain. This, however, does not appear to be the case in all brain regions. A recent study of striatal neurons in Fmr1 KO mice demonstrates an increase in basal inhibitory transmission driven by increased GABA release [Centonze et al., 2008]. This unique alteration is suggested to contribute to the frontostriatal dysfunction that is frequently observed in FXS patients [Hoeft et al., 2007; Menon et al., 2004], and may be involved in FXS-associated phenotypes such as attention deficit hyperactivity disorder and repetitive behaviors [Reiss et al., 1995]. As further evidence of the bidirectional nature of inhibitory alterations in Fmr1 KOs, recent evidence suggests increased numbers of inhibitory synapses in the CA1 region of the hippocampus [Dahlhaus and El-Husseini, 2010].
Together, studies in the amygdala, striatum, and subiculum reveal specific alterations in inhibitory transmission in Fmr1 KO mice, with those in the first two regions identifying clear presynaptic abnormalities and thus implicating dysfunction of presynaptic inhibitory interneurons as a key component of circuit defects. It is important to keep in mind, however, that normal inhibitory circuit function not only requires that interneurons form appropriate synaptic contacts onto their targets, as well as synthesize, release, and metabolize GABA in a dynamic way, but also that they receive appropriate excitatory input and respond normally to network activity. A study of parvalbumin-expressing fast-spiking (FS) interneurons in primary somatosensory (barrel) cortex and their integration with regular-spiking (RS) excitatory neurons reveals normal FS to RS transmission in Fmr1 KOs, but deficits in RS to FS transmission [Gibson et al., 2008]. This was found to be accompanied by an increase in the duration of stimulus-induced coordinated increases in network activity (UP states), as well as a decrease in gamma-frequency synchronization, illustrating important downstream effects of impaired activity-dependent interneuron activation at the cortical circuit level. These inhibitory circuit defects in Fmr1 KOs provide evidence in line with common FXS phenotypes such as cognitive impairment, sensory hypersensitivity, epilepsy, and abnormal cortical EEG patterns [Berry-Kravis, 2002; Berry-Kravis et al., 2010; Hagerman and Stafstrom, 2009; Miller et al., 1999; Musumeci et al., 1988, 1999] that likewise suggest excitatory/inhibitory imbalances in the cerebral cortex.
In addition to these alterations in basic inhibitory neurotransmission, two recent studies implicate aberrant modulation of transmission in the pathogenesis of FXS. GABA release is modulated by signaling through a number of presynaptic receptors, including the GABAB and cannabinoid receptors, which act to reduce GABA release [Bacci et al., 2005; Misgeld et al., 1995; Wilson and Nicoll, 2002]. In the CA1 region of the hippocampus, there is enhanced coupling of group I metabotropic glutamate receptor (mGluR) signaling and endocannabinoid (eCB) mobilization in Fmr1 KOs, which results in enhanced suppression of inhibitory transmission [Zhang and Alger, 2010]. This increase in the suppression of inhibition is proposed as a potential contributor to the enhanced mGluR-dependent synaptic plasticity previously observed in the Fmr1 KO hippocampus [Huber et al., 2002] and cognitive impairment in FXS. This finding not only suggests a link between excessive group I mGluR signaling and hyperplasticity of excitatory synapses in FXS, but also between mGluRs and alterations in inhibitory function, an important consideration, given that group I mGluRs are differentially expressed in a number of inhibitory neuron subtypes [Kerner et al., 1997; Stinehelfer et al., 2000; Sun et al., 2009]. Interestingly, excessive group I mGluR-eCB-dependent suppression of inhibition has also been observed in the striatum [Maccarrone et al., 2010], indicating that disruption of this signaling pathway may be disrupting inhibitory transmission in other brain regions as well.
It is not yet known whether other signaling pathways affecting GABA release, such as presynaptic GABAB receptor-mediated transmission, are similarly enhanced or disrupted in Fmr1 KOs. Given the evidence for altered GABAergic transmission and presynaptic function in a number of brain regions, the presynaptic GABAB receptors present an intriguing candidate for future studies.
Together, these findings from different key brain regions demonstrate that perturbations of inhibitory transmission are a hallmark of FMRP loss. These changes have distinct implications for the balance of excitation and inhibition, and thus, network function and behavioral output. In some ways, these inhibitory defects can be viewed as an ‘end result’ of disrupted circuit development. Little is known, however, about how these significant alterations arise over the course of brain development. Interneuron development and the formation and refinement of precise inhibitory circuits are highly complex and still incompletely understood. Nevertheless, it is within the framework of these processes that the inhibitory deficits observed in FXS patients and Fmr1 KO mice are presumed to arise. An understanding of the developmental timeline of the GABAergic system is therefore required in order to gain insight into the developmental mechanism of any neurodevelopmental disorder involving synaptic and circuit dysfunction. In the following section, we provide a brief outline of the role of GABA in development and the timeline of inhibitory circuit formation. We discuss how the observed molecular changes in the GABAergic system might drive alterations in mature neurotransmission, and vice versa. In doing so, we identify developmental processes that might be perturbed in the FXS brain.
GABA in the Development of Functional Neural Circuits
The development of functional inhibitory circuitry is a complex and prolonged process, extending from embryonic stages well into the postnatal period, that relies on a number of intrinsic and extrinsic factors, including gene expression, cell location, and exposure to neurotransmitters and neurotrophins [Batista-Brito and Fishell, 2009; Wang and Kriegstein, 2009]. The importance of FMRP for normal circuit development is illustrated by the fact that Fmr1 KO mice exhibit a number of defects suggestive of aberrant or delayed development, including disruptions in synaptic plasticity [Desai et al., 2006; Huber et al., 2002; Li et al., 2002; Suvrathan et al., 2010; Wilson and Cox, 2007], early, transient defects in cortical excitatory connectivity and cell morphology [Bureau et al., 2008], delayed timing of critical period plasticity [Harlow et al., 2010], increased cellular excitability [Chuang et al., 2005], and elevated numbers of immature dendritic spines [Comery et al., 1997; Galvez et al., 2003; Irwin et al., 2001].
The prevalence of inhibitory dysfunction in a neurodevelopmental disorder like FXS prompts an important question: do early GABAergic defects contribute to abnormal circuit development, or do these defects arise as a result of the influences of an abnormally developing circuit? The short answer to this question is that given the current data, we cannot be sure. The expression of FMRP begins at early embryonic stages in the mouse brain and persists throughout development and into adulthood [Bakker et al., 2000; Hinds et al., 1993; Wang et al., 2004]. A similar expression profile is observed in humans, with Fmr1 and FMRP detectable in the brain at the embryonic, fetal, and adult stages [Abitbol et al., 1993; Agulhon et al., 1999; Tamanini et al., 1997]. FMRP is therefore temporally well-positioned to regulate key steps of inhibitory circuit development, which include migration and fate specification, morphological development and formation of initial synaptic contacts, and synaptic refinement and circuit maturation. GABA signaling has a crucial role in all of these processes [Ben-Ari, 2002; Wang and Kriegstein, 2009], and given that normal GABAergic system function depends on the presence of FMRP, it is likely that GABAergic dysfunction during development contributes to mature circuit alterations in FXS. Analysis of forebrain expression of GABAergic synapse proteins in Fmr1 KO mice suggests a system that is at least partially compromised as early as P5 [Adusei et al., 2010]. Furthermore, the inhibitory circuit defects observed in the somatosensory cortex were observed as early as postnatal week 2 [Gibson et al., 2008]. Together, these are indicative of the presence of functional inhibitory deficits during key times in inhibitory circuit development. In light of this evidence for early depression of the GABAergic system, we focus this section on the known roles of GABAergic signaling in development.
In mature neurons, GABA’s action through GABAA receptors is inhibitory. Intracellular Cl– levels are low, and thus, upon channel opening, Cl– ions enter the cell, hyperpolarizing the membrane and reducing cellular excitability. During embryonic and early postnatal development, however, GABA is depolarizing and thus excitatory [Ben-Ari, 2002], due to a high intracellular Cl– concentration, maintained by the Na+/K+/Cl– co-transporter NKCC1 [Yamada et al., 2004]. Before the emergence of functional synapses in the brain, the depolarizing action of GABA exerts prominent effects on neuronal development, regulating the proliferation of neuronal progenitors [Haydar et al., 2000; LoTurco et al., 1995], as well as providing both permissive and stop signals for neuronal migration via GABAA and GABAB receptor signaling [Behar et al., 1998, 2000, 2001; López-Bendito et al., 2003; Manent et al., 2005]. GABAergic signaling, therefore, is an important determinant of early neuronal development, helping to set the stage for circuit formation. Interestingly, FXS patients exhibit structural alterations in a number of brain regions, including the caudate nucleus, amygdala, and hippocampus [Gothelf et al., 2008; Reiss et al., 1994], indicating that perturbations of proliferation and migration may indeed occur in the absence of FMRP. This issue remains largely unexplored in Fmr1 KO mice, however. Studies reveal no alterations in the gross morphology or in cell number in the basolateral amygdala of Fmr1 KOs at P21, and even more specifically, no changes in the number of GABA-expressing cells [Olmos-Serrano et al., 2010]. A study of adult Fmr1 KO mice, however, reveals slight changes in the distribution and density of PV-positive cells in primary somatosensory cortex [Selby et al., 2007], illustrating that specific neuronal populations might be sensitive to proliferative or migrational defects in FXS.
Provided that GABAergic neurons arrive at the correct locations, they are well-positioned to guide early circuit formation and maturation, first through the continued depolarizing action of GABA, then later through its inhibitory hyperpolarizing action. In the rodent hippocampus, functional GABAergic synapses are present on both pyramidal neurons and interneurons at birth, and are established before functional glutamatergic synapses [Hennou et al., 2002; Tyzio et al., 1999]. At this stage, GABA is still depolarizing, and acts through GABAA receptors to increase Ca2+ influx through NMDA receptors and promote glutamatergic synapse maturation [Akerman and Cline, 2006; Wang and Kriegstein, 2008]. In the absence of GABA’s early depolarizing action, these key processes are disrupted and result in imbalances in excitatory and inhibitory transmission and disrupted dendritic maturation [Akerman and Cline, 2006; Wang and Kriegstein, 2008]. Thus, GABA signaling is required at even the earliest stages of functional circuit maturation. As noted above, the Fmr1 KO brain exhibits abnormalities in glutamatergic synaptic function, including enhanced NMDA/AMPA ratios at early postnatal ages [Harlow et al., 2010] that may reflect developmental delays in synaptic maturation. It is not yet clear how GABAergic defects might contribute to alterations in glutamatergic synaptic function in FXS, but they provide an intriguing mechanistic candidate.
Inhibitory synaptogenesis is characterized by a period of rapid increase in synapse number and maturation that terminates around the end of the 4th postnatal week [De Felipe et al., 1997; Micheva and Beaulieu, 1996]. Not surprisingly, this process is also driven by GABAergic signaling. In the absence of neuronal activity or the rate-limiting GABA-synthesizing enzyme GAD67, inhibitory synaptogenesis is profoundly reduced [Chattopadhyaya et al., 2004, 2007]. Interestingly, inhibitory synaptogenesis can also be driven with GABAA and GABAB receptor agonists [Chattopadhyaya et al., 2007], illustrating a strong link between presynaptic activity, GABA production/release, and synaptic maturation. These observations in the mouse neocortex are intriguing in light of the findings that both GAD levels and inhibitory synapse numbers are disrupted in multiple brain regions of Fmr1 KO mice. In the context of the basolateral amygdala, which exhibits significant reductions in both of these measures in Fmr1 KO mice during the 4th postnatal week (see above), one clear hypothesis would be that blunted GAD levels – and therefore reduced GABA release and receptor activation – could be occurring early in development and causing reductions in inhibitory synaptic innervation of excitatory cells. In contrast, increased GABAergic synapse numbers [Dahlhaus and El-Husseini, 2010], as well as increased GAD levels [El Idrissi et al., 2005] are reported in the hippocampus, suggesting that early misregulation of GABA production might drive region-specific alterations in mature inhibitory function. The developmental timeline of GAD expression (GAD67 in particular) in Fmr1 KOs remains to be determined, but could provide a useful link between molecular and functional circuit alterations.
Studies in rodents and nonhuman primates indicate that the expression of GABAA receptor subunits is developmentally regulated in accordance with GABAergic synapse maturation [Fritschy et al., 1994; Golshani et al., 1997; Huntsman et al., 1999; Laurie et al., 1992]. This maturation generally involves the upregulation of subunits mediating ‘fast’ GABAA responses (fast IPSC decay kinetics) such as α1, and the concurrent downregulation of ‘slow’ subunits, such as α3 [Bosman et al., 2002; Dunning et al., 1999; Heinen et al., 2004; Hollrigel and Soltesz, 1997; Huntsman and Huguenard, 2000; Vicini et al., 2001], and thus comprises an important developmental ‘switch’. Notably, this subunit switching occurs alongside synaptogenesis [De Felipe et al., 1997] and the maturation of GABAergic transmission from excitatory to inhibitory, which similarly depends on a developmental switch – the upregulation of the Cl–-extruding K+/Cl– co-transporter KCC2 [Rivera et al., 1999; Wang et al., 2002]. The temporal coincidence of these processes indicates that they are all likely to be mechanistically related. Indeed, changes in IPSC decay mediated by the increase in synaptic α1 subunits are accompanied by changes in IPSC frequency and amplitude, as well as presynaptic release probability [Huntsman et al., 1999; Kirischuk et al., 2005; Kobayashi et al., 2008]. This presynaptic maturation is altered in α1 KO mice [Bosman et al., 2005; Ortinski et al., 2004], illustrating the requirement for correct postsynaptic receptor composition for synaptic maturation. In addition, inhibitory synapse stability has been shown to depend on GABAA receptor clustering by the scaffolding protein gephyrin [Fritschy and Brunig, 2003; Li et al., 2005; Yu et al., 2007], as well as by neuroligin-2 (NL-2)-neurexin interactions [Chih et al., 2005; Chubykin et al., 2007; Gibson et al., 2009; Hines et al., 2008; Poulopoulos et al., 2009; Varoqueaux et al., 2004]. Interestingly, FMRP can associate with NL-2 mRNA and influence expression of the NL-2 protein [Dahlhaus and El-Husseini, 2010], and genetic manipulation of NL-2 levels in mice leads to imbalances in excitatory and inhibitory transmission, anxiety-related behaviors, stereotypies, and abnormal social behaviors consistent with models of ASD and FXS [Blundell et al., 2009; Hines et al., 2008; Moy et al., 2009], an intriguing finding given the known associations of other neuroligin isoforms with ASD [Südhof, 2008].
An important consideration when investigating neuronal and circuit function in Fmr1 KOs is that, of the numerous GABAergic synapse components with altered expression, direct binding has only been shown for the GABAA receptor δ subunit. The changes in the expression of other receptor subunits or GAD, for example, might therefore arise not due to a loss of their direct interactions with FMRP, but instead could reflect secondary effects of altered mRNA stability [De Rubeis and Bagni, 2010] or changes in intercellular communication resulting from translational misregulation of other FMRP-binding mRNAs. Alternatively, these widespread changes in the GABAergic system could constitute compensatory changes in response to abnormalities in the developing circuit, such as an altered balance of excitation and inhibition. As an example, reductions in inhibitory synapse number in the striatum are proposed to arise in order to offset an increase in GABA release [Centonze et al., 2008]. Along the same lines, increases in IPSC decay kinetics in the basolateral amygdala [Olmos-Serrano et al., 2010] might reflect a homeostatic upregulation of ‘slow’ GABAA receptor subunits in response to reductions in inhibitory input, with increased IPSC duration acting to compensate for decreases in IPSC amplitude and frequency. Homeostatic changes in synaptic inhibition are frequently observed in sensory cortex in response to changes in circuit activity [Li et al., 2009; Maffei and Turrigiano, 2008; Maffei et al., 2006; Micheva and Beaulieu, 1995] and represent an important form of network plasticity that is likely to emerge in a number of neurodevelopmental disorders.
Currently, it is not entirely clear whether the circuit deficits observed in Fmr1 KOs represent developmental delay, homeostasis, or a wholly aberrant circuit. It is clear, however, that the potential developmental implications of a dysfunctional GABAergic system are numerous and complex. At the moment, detailed developmental studies of the GABAergic system in Fmr1 KOs are largely lacking, and studies, for example of GABAA receptor function – given the pronounced disruptions in expression, would be highly informative in this respect. Still, it is important to keep in mind that normal circuit development requires the actions of several dynamic intrinsic and extrinsic factors, many of which are likely contributors to the pathogenesis of FXS, including, for example, neurotrophic factors. Given that there is currently little data regarding these factors in the FXS brain, we have not focused on this issue. It is noteworthy, however, that semaphorin 3F, which can act as a chemorepellent, guide the migration [Marín et al., 2001; Tamamaki et al., 2003] and axon outgrowth [Pascual et al., 2005] of GABAergic interneurons, has been identified as a binding partner of FMRP [Darnell et al., 2001].
GABAergic System Components as Potential Therapeutic Targets for FXS
To date, a primary therapeutic target in FXS has been the metabotropic glutamate receptor 5 (mGluR5), based on evidence of unregulated signaling downstream of this receptor in Fmr1 KO mice [Chuang et al., 2005; Dolen and Bear, 2008; Jacquemont et al., 2011; Yan et al., 2005]. Given the increasingly understood prevalence of inhibitory dysfunction in FXS, however, the GABAergic system also presents a number of relevant and intriguing targets for treatment that are distinct from therapeutics aimed at reducing mGluR signaling. As described above, current evidence generally reveals an extensive dampening of GABAergic function throughout the adult FXS brain, and this dysfunction has been implicated in many of the hallmark symptoms of FXS, including anxiety, autistic behaviors, epilepsy, and cognitive impairment. GABAergic compounds, in turn, have demonstrated therapeutic efficacy in many of these disorders. Therefore, compounds targeting the GABAergic system may provide novel, effective, and in some cases, currently available treatment options for the symptoms of FXS (fig. 3; table 2).


Potential pharmacological targets for the treatment of FXS. Numbers identify loci of action of pharmacological compounds targeting components of the GABAergic system (inset legend; table 2). Note: The presumed actions of arbaclofen (4) are illustrated as predominately postsynaptic, since presynaptic modulation is expected to reduce GABA release via a reduction in voltage-dependent calcium influx. However, since glutamatergic terminals also express presynaptic GABAB receptors, additional beneficial actions of arbaclofen could also lie in the reduction of glutamate release from excitatory synapses.
Potential pharmacological targets for the treatment of FXS. Numbers identify loci of action of pharmacological compounds targeting components of the GABAergic system (inset legend; table 2). Note: The presumed actions of arbaclofen (4) are illustrated as predominately postsynaptic, since presynaptic modulation is expected to reduce GABA release via a reduction in voltage-dependent calcium influx. However, since glutamatergic terminals also express presynaptic GABAB receptors, additional beneficial actions of arbaclofen could also lie in the reduction of glutamate release from excitatory synapses.
The first study that systematically identified GABAergic components as possible targets for FXS treatment used the Fmr1 KO Drosophila melanogaster model of FXS to screen 2,000 compounds for their ability to rescue glutamate-induced toxicity in developing fly larvae related to hypertrophic mGluR signaling [Chang et al., 2008]. Of the 9 compounds identified in this screen, 3 acted on the GABAergic system: nipecotic acid (a GAT blocker), creatinine (a GABAA receptor activator), and GABA itself. Not only did these compounds rescue glutamate-induced lethality, but they also rescued characteristic cellular, morphological, and behavioral phenotypes related to FXS in the adult Fmr1 KO flies. Therefore, compounds that increase the function of the GABAergic system, either by increasing the availability of GABA (nipecotic acid) or acting directly at GABA receptors (creatinine and GABA), can rescue FXS phenotypes.
Two clinically available, FDA-approved anticonvulsants function like nipecotic acid to increase the availability of GABA and therefore could show efficacy in FXS. The first, tiagabine, specifically blocks the presynaptic GABA transporter 1 (GAT1), thus increasing synaptic GABA levels and enhancing phasic and tonic GABAergic inhibition [Nielsen et al., 1991]. The second, vigabatrin, blocks the catabolism of GABA by inhibiting the function of GABA transaminase (GABA-T), which is required for the breakdown of GABA to glutamate. This blockade increases GABA availability, both intracellularly, for packaging into presynaptic vesicles, and extracellularly [Chiron et al., 1997; French et al., 1996]. Although these compounds exhibit efficacy at improving GABAergic function, they do not work in all patients and have a high incidence of side effects, including retinal neuropathy [Frisén and Malmgren, 2003]. Their GABAergic actions, however, may warrant investigation in FXS.
Many compounds act directly on GABAA receptors, and therefore may also improve symptoms of FXS. For instance, the free amino acid taurine acts as an agonist at GABAA receptors, increases GAD expression, increases GABA levels, induces changes in GABAA receptor subunit composition [L’Amoreaux et al., 2010], and decreases seizure susceptibility [El Idrissi and L’Amoreaux, 2008]. This endogenous amino acid is developmentally dysregulated in Fmr1 KO mice [Gruss and Braun, 2004], and chronic taurine feeding improves cognition [El Idrissi et al., 2009] and neuroendocrine symptoms in these animals [El Idrissi et al., 2010].
Several other compounds that potentiate GABAA receptor function have been identified and/or examined in preclinical and clinical trials for FXS or its related symptoms. One compound, riluzole, has multiple modes of action to decrease excitability, including GABAA receptor potentiation [Jahn et al., 2008] and GAT blockade [Mantz et al., 1994]. Riluzole is an FDA-approved anticonvulsant often utilized for amyotrophic lateral sclerosis treatment in adults and in one preclinical trial shows mild efficacy in improving attention deficit disorder with hyperactivity in adult FXS patients [Erickson et al., 2010]. Another compound, gaboxadol, acts preferentially at extrasynaptic, tonic δ-subunit-containing GABAA receptors to dampen cellular excitability via increased tonic inhibitory conductance [Brown et al., 2002; Glykys and Mody, 2007]. This compound, previously used as a sleep aid [Deacon et al., 2007; Lundahl et al., 2007], rescues the hyperexcitability of principal output excitatory neurons in the basolateral amygdala of Fmr1 KO mice [Olmos-Serrano et al., 2010], clearly demonstrating its potential therapeutic benefit in FXS patients, particularly with respect to amygdala-based behaviors, such as anxiety and features of ASD.
Benzodiazepines are proven, effective GABAA receptor agonists, but often present unwanted side effects, including sedation and rebound symptoms such as anxiety when treatment is discontinued [Nemeroff, 2003], and therefore might not be the optimal agents to treat symptoms of FXS. Interestingly, another class of molecules, the neurosteroids, increase GABAergic receptor function via positive allosteric modulation [Belelli and Lambert, 2005]. Natural neurosteroids such as allopregnanolone are not orally active, but one new synthetic neurosteroid, ganaxolone, the 3β-methyl analogue of allopregnanolone, is orally active, lacks hormonal side effects, and has entered phase II clinical trials for infantile spasms, partial seizures, and catamenial epilepsy [Reddy, 2010]. A neuroactive steroid like ganaxolone may be useful in the treatment of anxiety and seizures associated with FXS, and has advantages over benzodiazepines, given the low occurrence of side effects and evidence of preferential effects at extrasynaptic, tonically active δ-subunit-containing receptors [Biagini et al., 2010; Mihalek et al., 1999; Reddy, 2010].
Finally, GABAergic function can also be enhanced via the activation of postsynaptic GABAB receptors, and stimulation of these receptors has shown promise as a treatment in animal models of FXS. Although no published data describes postsynaptic GABAB currents in Fmr1 KO mice, activation of these receptors with the agonist baclofen reduces audiogenic seizure susceptibility in these animals [Pacey et al., 2009]. Accordingly, phase II clinical trials for safety and efficacy of the most active isomer of baclofen, arbaclofen, are currently being conducted in FXS patients (clinicaltrials.gov). The use of GABAB receptor-targeting therapeutics, however, could be complicated by any actions on presynaptic receptors, which act to reduce GABA release (and thus decrease synaptic inhibition) via inhibition of voltage-gated Ca+ channels [Padgett and Slesinger, 2010].
Concluding Remarks
FXS is a pervasive neurodevelopmental disorder characterized by extensive synaptic and circuit dysfunction, and profound alterations in the GABAergic system are now emerging as major contributors to the hyperexcitable circuit phenotype. Studies in Fmr1 KO mice reveal disturbances of the GABAergic system and functional inhibitory neurotransmission in a number of brain regions that are highly relevant to the FXS phenotype, including the amygdala, cerebral cortex, hippocampus, and striatum. Thus, the GABAergic system presents an important pharmacological target for the treatment of a number of the neurological manifestations of FXS.
While GABAergic dysfunction has been implicated in a number of disease states involving synaptic and circuit dysfunction, the similarities between FXS and other developmental disorders are particularly intriguing. Rett syndrome, for example, is similarly characterized by intellectual disability and elevated incidences of ASD [Moretti and Zoghbi, 2006], and mouse models of Rett syndrome, like FXS models, exhibit prominent alterations in GABAergic function [Chao et al., 2010; Dani et al., 2005; Medrihan et al., 2008]. This parallel between FXS and Rett syndrome models suggests that GABAergic dysfunction may represent a common pathway to brain dysfunction in neurodevelopmental disorders.
The development and maturation of neural circuits relies on a series of highly precise and complex processes, and disturbances of these processes can drastically alter mature circuit function. GABAergic signaling is essential for correct neuronal migration, maturation, and circuit formation, and defects in the GABAergic system are therefore likely to have profound effects on neuronal development and circuit function in FXS. Currently, early developmental alterations in GABAergic function are not well understood in FXS, but an understanding of these should provide important insight into the nature of the FXS brain, as well as valuable information about key pharmacological targets. It is important to keep in mind, however, that the role of GABA in the developing CNS is dynamic and variable between brain regions (i.e. cortex, amygdala, striatum). Therefore, the same GABAergic effectors that aid adult patients could have adverse effects in developing individuals based on the role of GABA in particular brain regions at particular developmental time periods. In order to effectively utilize GABAergic targeting for FXS treatment, we must understand more about the role of GABA in specific brain regions during development of the FXS individual and develop specific treatments to affect region-specific circuit changes. Despite the challenges, further study of the GABAergic system will reveal crucial details of the developmental pathology of FXS as well as unique, targeted treatments for the disease.
Acknowledgements
This work was supported by NIH/NINDS (R01NS053719), Autism Speaks, and FRAXA grants to M.M.H., a CIHR doctoral research award to S.M.P., and an Epilepsy Foundation predoctoral training fellowship to B.S.M.
