


Although it is known that insulin-dependent (type 1) diabetes results in depressed contractile performance associated with diminished sarcoendoplasmic reticular Ca2+-ATPase (SERCA2a) activity, findings in insulin-resistant (type 2) diabetes suggest a less clear association. The db/db insulin-resistant mouse model exhibits decreased cardiac performance both in situ and in isolated ex vivo working hearts. In this study, contractile performance and calcium transients were measured in Langendorff-perfused hearts and isolated cardiac myocytes. Diabetic (db/db) mouse hearts demonstrated decreased rates of contraction, relaxation, and pressure development. Calcium transients from isolated myocytes revealed significantly lower diastolic and systolic levels of calcium in diabetic hearts. Furthermore, the decay rate of the calcium transient was significantly reduced in diabetic myocytes, suggesting a diminished capacity for cytosolic calcium removal not associated with a change in sodium-calcium exchanger activity. Calcium leakage from the sarcoplasmic reticulum (SR) measured using tetracaine was significantly increased in diabetic myocytes. Western blot analysis indicated only a small decrease in SERCA2a expression in diabetic mice, but a large increase in phospholamban expression. Expression of the ryanodine receptor did not differ between groups. In conclusion, the decreased contractile function observed in the db/db diabetic mouse model appears to be related to decreased calcium handling by the SR.
Heart disease is the leading cause of death among diabetic patients. It has been recognized for a number of years that diabetes can impair myocardial performance independent of coronary artery disease or hypertension (1,2). Type 2 (non-insulin-dependent) diabetes is the most prevalent form of this disease, resulting from a combination of insulin resistance (resulting in hyperinsulinemia) and ultimately a failure of pancreatic β-cells to maintain adequate insulin secretion to overcome resistance (3,4). The etiology of type 2 diabetes is complex, arising from a mixture of genetic and environmental factors. In contrast, type 1 (insulin-dependent) diabetes is characterized by decreased insulin secretion and is readily reversed by insulin replacement. Although a number of studies have examined the effect of type 1 diabetes on contractile function in the heart, fewer such studies have been performed in type 2 diabetic models.
The db/db mouse model of type 2 diabetes was originally isolated from the C57BLK6 strain >30 years ago (5). Disease development in this strain follows a distinct pattern. Outwardly, the mice are indistinguishable from their nondiabetic littermates until soon after weaning, when they show an accelerated gain in body mass. Initially, insulin secretion increases to overcome insulin resistance in the periphery to prevent hyperglycemia. By age 8–12 weeks, the maximum extent of hyperinsulinemia is attained; hyperglycemia then develops when the insulin levels are no longer sufficient to counter the insulin resistance (6,7). In the diabetic state, these mice show a diminished capacity for cardiac power output in the isolated working heart preparation (8). This effect is age dependent with the onset of diabetes (9), an observation confirmed in vivo by echocardiography (10). Although this model shows some characteristics similar to type 1 diabetes, including altered metabolic substrate preference (11), it is not known how calcium handling is affected in this model.
Alterations in myocyte calcium handling in type 1 diabetes have been well documented (12–14). The type 1 diabetic model is associated with a depression in contractile function underlined primarily by reduced sequestration of calcium into the sarcoplasmic reticulum (SR) as a result of decreased sarcoendoplasmic reticular Ca2+-ATPase (SERCA) pump activity. The decreased SERCA pump activity is the result of either a decreased expression of SERCA itself or an increase in the prevalence of its inhibitory protein phospholamban (PLN) (15,16). In contrast, relatively few studies on contractility have been performed on models of type 2 diabetes. The JCR:LA-cp rat exhibits severe insulin resistance; however, studies with this model have resulted in a series of contradictory results. Isolated working hearts from cp/cp rats have shown a rapid decline in function unless large concentrations of insulin are added to the perfusion medium and the perfusate calcium concentration is reduced (17). Under these conditions, no functional difference has been observed between cp/cp rats and their lean littermates. In contrast, Langendorff-perfused cp/cp hearts have exhibited a modest reduction in contractility at age 3 months, but not at older ages (18). Despite this observation, Misra et al. (19) actually found an increase in calcium uptake in isolated SR fractions in 3-month-old cp/cp rats. Studies examining contractility in ZDF rats have shown either no change, a slight increase, or a decrease in contractile function (20–22). Because of the profound depression in cardiac function obtained from db/db mice relative to other models, we decided to examine myocardial contractility and calcium handling in these mice.
RESEARCH DESIGN AND METHODS
This investigation conformed to the Guide for the Care and Use of Laboratory Animals as published by the National Institutes of Health (NIH Publ. no. 85-23, revised 1996).
Male C57BL/KsJ-leprdb/leprdb diabetic (db/db) and nondiabetic littermate C57BL/KsJ-leprdb/lepr+ (db/+) mice, age 8 weeks, were obtained from The Jackson Laboratories (Bar Harbor, ME) and housed with ad libitum food and water for an additional 4 weeks until age 12 weeks. Their final body mass was determined just before they were killed, and blood was collected from the chest cavity after the heart was excised. The increased body mass and blood glucose (fed state) of the db/db mice confirmed their diabetes status (Table 1).
Langendorff-perfused hearts.
Animals were killed by an overdose of pentobarbital, and their hearts were excised from the chest. The aorta was cannulated, and perfusion was initiated with a modified Krebs-Henseleit buffer at a perfusion pressure of 55 mmHg. The Krebs-Henseleit buffer was gassed continuously with 95% O2/5% CO2 (37°C) and consisted of (in mmol/l): NaCl 118, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25.0, Na2EDTA 0.5, glucose 11, Na-pyruvate 0.5, and Na-octanoate 0.4. Octanoate was added to the perfusion medium to provide a readily metabolizable short-chain fatty acid because of the increased preference of db/db mice for ATP derived from fatty acids (8). A small balloon was made from polyethylene and inserted into the left ventricle through the pulmonary vein (16). The balloon was inflated to an end diastolic pressure of 10 mmHg. Pressure development was recorded digitally (1 kHz) by connecting the intraventricular balloon to a 2 F Millar pressure transducer. Hearts were lowered into a water-jacketed chamber (37°C) to maintain the temperature. The hearts were paced at 400 bpm and the resulting pressure waves were analyzed for pressure derivatives (+dP/dt, −dP/dt) and pressure development.
Individual myocyte measurements.
Individual myocytes were isolated from the excised hearts by collagenase digestion according to the method outlined by Belke et al. (23). Calcium transients were measured in myocytes affixed to laminin-coated coverslips and loaded with the fluorescent calcium indicator indo 1 (acetoxymethyl ester), according to the methods outlined by Suarez et al. (24). After being loaded, calcium transients were measured using a Solomere Technologies (Salt Lake City, UT) system configured for dual emission, attached to a Nikon Diaphot epifluorescence microscope, and recorded at 100 Hz using the Mac Lab (AD Instruments, Colorado Springs, CO) computer acquisition system. During these measurements, myocytes were constantly perfused with a HEPES-buffered Tyrode solution (pH 7.4) containing 1 mmol/l Ca2+, and electrically paced at a rate of 0.3 Hz. All myocyte experiments were carried out at room temperature. Leakage measurements were obtained by perfusing myocytes with 1 mmol/l tetracaine followed by rapid switching to 10 mmol/l caffeine, as outlined by Shannon et al. (25). Sodium-calcium exchanger (NCX) activity was assessed by first perfusing myocytes in the presence of 10 mmol/l caffeine (to inhibit SR calcium uptake) and recording the transient, then recovering contractility of the myocyte, and subsequent reperfusing them in a 0 Na−, 0 Ca−free 10 mmol/l Tyrode buffer (containing Li and EGTA instead of Na+ and Ca2+) with caffeine to inhibit NCX activity. A linear rate of decay for the caffeine-induced calcium transient was calculated using the first 3 s (300 time points) after the initial upstroke of the transient. The difference in the decay rate of the caffeine-induced calcium transient between the two traces was taken to represent NCX activity (26) (Fig. 1A).
Western blot analysis.
Ventricular tissue was homogenized using a Polytron homogenizer. After protein content of the homogenate was measured (Bradford Protein Assay; Bio-Rad, Hercules, CA), protein separation was achieved by resolving 20 μg protein (unless otherwise specified) on a 4–20% Bis-Tris polyacrylamide gel (Invitrogen, Carlsbad, CA) and transferring the protein to a nitrocellulose membrane. The nitrocellulose membrane was blocked overnight at 4°C using 5% nonfat milk powder in Tris-buffered saline (+0.05% Tween 20) and exposed for 2 h to primary antibodies directed against SERCA2a (rabbit polyclonal, made in the laboratory), phospho-(serine-16)-phospholamban (rabbit polyclonal; Upstate Biotechnology), PLN (mouse monoclonal; Affinity Bioreagents), ryanodine receptor (RyR) 2 (mouse monoclonal; Affinity Bioreagents), and the FK506 binding protein FKBP (goat polyclonal; Santa Cruz Biotechnology). The blots were then washed, exposed to secondary antibodies conjugated to horseradish peroxidase to drive a chemiluminescence reaction (Amersham, Little Chalfont, U.K.), and exposed to film. Band densities were quantified with the Scion Image Software for personal computers (Scion, Frederick, MD).
Statistics.
Data are expressed as means ± SE. Comparisons between the diabetic and control groups were performed using an unpaired t test.
RESULTS
Contractile function in db/db hearts.
Cardiac function in ex vivo Langendorff-perfused hearts was measured using a fluid-filled intraventricular balloon. Pressure development was significantly lower in diabetic than in control mice (123 ± 3 vs. 150 ± 7 mmHg), suggesting a diminished capacity for force generation (Fig. 2C). Similarly, the rates of pressure development (+dP/dt) and relaxation (−dP/dt) were both significantly lower in diabetic mice (Fig. 2A and B). This latter observation suggests a possible defect in SR calcium uptake through SERCA2a.
Calcium transients in myocytes from db/db hearts.
To examine calcium handling in db/db mice, myocytes were isolated by collagenase digestion, plated on laminin-coated coverslips, and loaded with the calcium-sensitive dye indo 1. Representative calcium transients from diabetic and control mice are shown in Fig. 3A. Resting diastolic calcium levels in diabetic myocytes were roughly half the values obtained from control myocytes (Fig. 3C). The absolute height of the calcium transient reached after stimulation was significantly larger (P < 0.05) in the control than in the diabetic group (210 ± 21 vs. 108 ± 15 nmol/l) (Fig. 3D). Furthermore, the half-time of calcium decay from systolic levels was significantly longer (average 33%) in diabetic than control myocytes (Fig. 3B), suggesting a slower removal of cytosolic calcium.
Sodium-calcium exchanger activity.
To determine if the slower decay of the calcium transient could be due to NCX activity, caffeine-induced transients were measured in normal Tyrode and 0 Na, 0 Ca Tyrode (Li, EGTA) solution, with the difference representing NCX activity. As shown in Fig. 1B, no difference in NCX activity could be observed between diabetic and control groups. This suggests that NCX activity was unlikely to have contributed significantly to the difference in the calcium transient observed between the two groups. The absolute extent of calcium released after exposure to 10 mmol/l caffeine was significantly higher in the control group, suggesting increased SR calcium loading (Fig. 1C).
Sarcoplasmic reticulum calcium leakage.
In addition to observing calcium transients in control and diabetic mice, we also examined SR calcium leakage. The addition of 1 mmol/l tetracaine to the perfusion medium blocked the release of calcium through the ryanodine channel, but permitted the uptake of calcium by SERCA2a. An example of the measured leakage is shown in Fig. 4A. After pacing (0.3 Hz) myocytes to obtain a baseline indo 1 ratio, the cells were perfused with a modified Tyrode (0 Na, 0 Ca) solution containing 1 mmol/l tetracaine to block calcium efflux through the RyR. Blocking the RyR led to a decrease in the indo 1 ratio corresponding to the calcium leakage from the SR. The total SR calcium load ([Ca]SRT) was subsequently determined by a brief (5-s) exposure of the myocyte to modified Tyrode (0 Na, 0 Ca) solution containing 10 mmol/l caffeine. The extent of calcium leaked from the SR was subsequently normalized to the total SR calcium load. Under these conditions, the leakage was estimated to be 12% in control mice and 32% in diabetic mice (Fig. 4B). The absolute extent of calcium leakage inhibited by tetracaine below baseline level was significantly higher in the diabetic group (38.1 ± 5.3 vs. 17.9 ± 2.9 nmol/l Ca2+; P < 0.05). Although the leakage was expected to increase with an increasing calcium load of the SR, the calcium load after exposure to tetracaine and release by caffeine was actually slightly lower in the diabetic than in the control group (146 ± 27 vs. 186 ± 21 nmol/l Ca2+).
Western blot analysis.
To determine if a difference in protein expression plays a role in the functional differences observed between control and diabetic hearts, tissue samples were homogenized and subjected to SDS-PAGE. Western blot analysis revealed only a small (NS) decrease in SERCA2a expression in diabetic hearts (Fig. 5). In contrast, expression of the regulatory protein PLN was significantly increased in diabetic hearts, so that the ratio of PLN/SERCA2a expression was approximately threefold higher in diabetic than in control mice. Analysis of the phosphoserine 16 form of PLN revealed a slight decrease (NS) in the diabetic group that may also have contributed to an increased inhibitory effect of PLN on SERCA2a. Analysis of RyR2 expression indicated only a small (NS) decrease in the diabetic group, indicating that the increased leakage observed in this group was not due to increased protein expression of the RyR. However, we did find a significant 24% decrease in the expression of FKBP 12.6 in the diabetic group, which could contribute to the increased leak.
DISCUSSION
In the present study, we examined contractility in Langendorff-perfused hearts and calcium handling in isolated myocytes from diabetic (db/db) and nondiabetic (db/+) mice. Consistent with previous findings, the increase in body weight and blood glucose in 12-week-old diabetic mice was accompanied by a depression in cardiac performance (8–10). A number of studies have established that type 1 diabetes leads to a decreased contractility in the heart associated with decreased calcium handling due to a depression in SERCA2a activity (12,13). We have previously demonstrated that increasing SERCA2a activity by protein overexpression improves contractile function in streptozotocin-induced type 1 diabetes (16). Myocardial contractility in type 2 diabetic models has not been explored as extensively, and the existing studies have had contradictory results. The development of insulin resistance in rats caused by a high-sucrose diet results in reduced myocyte contractility and calcium handling, even in the absence of an overt increase in plasma glucose (27,28). In contrast, JCR: La corpulent (cp/cp) rats are profoundly insulin resistant; however, although their hearts may show a modest decrease in contractility at age 3 months, this phenomenon is not apparent in older rats (18). Furthermore, Misra et al. (19) have reported an increase in SR calcium uptake from 3-month-old cp/cp rats. In 14-week-old ZDF rats, Ren et al. (21) found no difference in the characteristics of isolated myocyte shortening and relengthening when compared with lean controls, although they did note a slight decrease in diastolic calcium and a prolonged decay time for calcium transients from diabetic rats. They did not observe any difference in plasma glucose between the groups. Using 11-week-old ZDF rats, Chatham and Seymour (22) observed no difference in developed pressure or the rate-pressure product in Langendorff-perfused hearts, although obese animals had a plasma glucose level three times higher than that of their lean littermates. Young et al. (29) have shown a decreased cardiac power output of isolated working hearts from obese ZDF rats, and Zhou et al. (20) have reported a significant decrease in fractional shortening in obese ZDF rats measured in vivo by echocardiography. A similar echocardiographic study using db/db mice revealed an age-dependent decrease in fractional shortening with an increase in plasma glucose and the overt development of diabetes (10). A similar age-dependent decrease in cardiac function obtained from the isolated working heart preparation of db/db mice has also been observed (9).
In the present study, we observed a decrease in contractile function in 12-week-old diabetic (db/db) mice having an increased body weight and elevated blood glucose. Although a previous study examining the isolated working heart did not show a significant decrease in dP/dt (8), we did observe a significant decrease in both positive and negative dP/dt in the present study. This discrepancy may be attributable to the methods used to obtain the results. The previous study (8) used a work performing heart (doing both pressure and volume work), whereas our study used an intraventricular balloon (pressure work only). We observed not only a significant decrease in dP/dt in diabetic mice, but also a lower extent of pressure development. Because increased collagen deposition or an increase in glucose-mediated collagen cross-linking in db/db mice may affect contractile properties independent of myocyte pathology (30), we examined calcium handling in isolated cardiac myocytes. In support of the observations made in the whole heart, we observe that the calcium transients obtained from diabetic myocytes were smaller than those from the control group. Furthermore, the amount of calcium that could be released after caffeine stimulation was lower, suggesting a decreased SR calcium load. The decreased rate of decay of the calcium transient seen in the diabetic group matched the decreased rate of relaxation observed in whole hearts. This observation was likely due to differences in SR activity and not to an additional effect of sarcolemmal NCX activity, as NCX activity was similar in both groups. This latter observation differs from findings in type 1 diabetic models, where NCX activity is markedly decreased in the diabetic state (31,32), an observation we have confirmed in streptozotocin-administered National Institutes of Health Swiss mice using the same methods described in the present study (D.D.B., unpublished observations).
To find the underlying reason for the decreased SR calcium handling observed in the diabetic mice, we examined SR protein expression. In looking at the expression of SERCA2a, we observed only a small (NS) decrease in the diabetic state. This observation parallels one reported by Young et al. (29) in obese ZDF rats, where they also found only a small (NS) decrease in SERCA2a mRNA expression levels. We also observed a large increase in the expression of the SERCA2a inhibitory protein PLN in the diabetic group, which in conjunction with the small decrease in SERCA2a expression would increase the ratio of PLN/SERCA2a. A similar observation has been made in type 1 diabetic models (15). The phosphorylation status of PLN, especially at serine residue 16, can alter its ability to interact with SERCA2a. Previous studies have indicated a decrease in sympathetic nerve activity in the hearts of db/db mice (33), which could lead to decreased phosphorylation of PLN. In examining the phosphorylation status of serine 16, we observed only a small (NS) decrease in phospho-PLN in the diabetic group. However, given the large increase in PLN expression in the diabetic group, this modest decrease in phospho-PLN would indicate that a majority of the protein exists in the inhibitory nonphosphorylated state. This effect would increase the inhibitor action of PLN on SERCA2a activity.
Although a depression in calcium uptake would decrease SR calcium loading, we examined calcium leakage from the RyR to determine if this might also play a role. We observed a significant increase in leakage in the diabetic group, although RyR2 expression was slightly lower in that group. Although leakage through the RyR generally increases with increase SR calcium loading (25), in our study, SR calcium loading was lower in the diabetic than in the control group. The underlying reason for the increased leakage may relate to the decreased expression of FKBP 12.6 observed in the diabetic group. FKBP 12.6 is an accessory protein associated with the RyR, and plays a role in coordinating the opening and closing of individual RyRs in an array (34). The loss of FKBP 12.6 binding to the RyR increases the open probability of the receptor (35). Alternately, the increased leakage may have been related to modification of the receptor directly. Recently, Bidasee et al. (36) reported increased glycation of RyR2 in streptozotocin-induced diabetic rat hearts. Although the specific contribution of increased leakage to cardiac dysfunction in diabetes cannot be described, it may play a role in the decreased cardiac efficiency observed in type 2 diabetic models. Because calcium handling by the SR uses a significant portion of the ATP required for each heart beat, any leakage of calcium from the SR not being directed toward force generation by the myofibrils would decrease cardiac efficiency. Such a decrease in cardiac efficiency has been previously observed in db/db mice and ZDF rats (8,29).
The decreased contractile performance of the insulin-resistant (type 2) diabetic db/db model parallels the decreased contractile performance observed in insulin-deficient (type 1) diabetic models. Although plasma insulin levels differ between the two models, the db/db diabetic model shows profound hyperglycemia (as does the type 1 model) and exhibits a progressive cardiomyopathy with increasing plasma glucose levels and age (9). In contrast, many other insulin-resistant models without notable hyperglycemia may show only a modest cardiomyopathy (21), as do mice lacking a cardiac myocyte−specific insulin receptor; these mice also show no derangement in glucose homeostasis (23). In some profoundly insulin-resistant models without hyperglycemia, no decrease in contractile performance has been observed (19). These observations suggest that the development of hyperglycemia, if not directly responsible for the contractile phenotype, can certainly exacerbate it. To this end, we have observed the effect of high glucose in depressing SERCA2a expression in cardiac myocytes, likely acting through O-linked glycosylation of transcription factors (37). Such a role for hyperglycemia in mediating protein transcription in a type 2 diabetic model remains to be examined. In contrast, exposure of adult rat ventricular myocytes to high glucose slows SERCA2a activity without affecting protein expression (38).
In summary, we observed a decrease in contractile function and calcium handling in the db/db mouse heart that can be attributed to a decline in SR activity. In this respect, the db/db (type 2) diabetic model parallels observations made about the insulin-deficient type 1 diabetic model. As a result, therapeutic strategies aimed at improving SERCA2a activity (e.g., gene therapy) may be beneficial in type 2 diabetic cardiomyopathy.

Calcium transients in myocytes perfused with 10 mmol/l caffeine. A: An example of the calcium transient obtained from a myocyte perfused first with caffeine in normal Tyrode (Caf, NT) solution and later with caffeine in a modified Tyrode (Caf, 0 Na, 0 Ca) solution in which the sodium and calcium were removed to block NCX activity. The difference in decay between the two transients was taken to represent NCX activity (B), which was not significantly different between the control (n = 13 myocytes) and diabetic (n = 17 myocytes) groups. In contrast, the absolute height of the calcium transient (peak − base) stimulated by caffeine (C) was significantly lower in the diabetic group, indicating a reduced SR calcium load in this group. *P < 0.05.
Calcium transients in myocytes perfused with 10 mmol/l caffeine. A: An example of the calcium transient obtained from a myocyte perfused first with caffeine in normal Tyrode (Caf, NT) solution and later with caffeine in a modified Tyrode (Caf, 0 Na, 0 Ca) solution in which the sodium and calcium were removed to block NCX activity. The difference in decay between the two transients was taken to represent NCX activity (B), which was not significantly different between the control (n = 13 myocytes) and diabetic (n = 17 myocytes) groups. In contrast, the absolute height of the calcium transient (peak − base) stimulated by caffeine (C) was significantly lower in the diabetic group, indicating a reduced SR calcium load in this group. *P < 0.05.

Left ventricular pressure measurements obtained from Langendorff-perfused mouse hearts using an intraventricular balloon. The maximal rates of contraction (A) and relaxation (B) were significantly lower in the diabetic (db/db) than in the control group. Peak systolic pressure (C) was also significantly lower in the diabetic group (n = 6 hearts each group). *P < 0.05.
Left ventricular pressure measurements obtained from Langendorff-perfused mouse hearts using an intraventricular balloon. The maximal rates of contraction (A) and relaxation (B) were significantly lower in the diabetic (db/db) than in the control group. Peak systolic pressure (C) was also significantly lower in the diabetic group (n = 6 hearts each group). *P < 0.05.

Indo 1 calcium transients obtained from isolated cardiac myocytes. A: An example of the transients obtained from control and diabetic mice indicating the smaller transient observed in the diabetic group. Analysis of the decay of the transient from peak calcium values (B) revealed a longer decay time in the diabetic group. Measurement of diastolic (C) and absolute transient height (D) calcium levels indicated that both values were significantly lower in the diabetic group, with values being roughly 50% of those measured in the control group. Data represent means ± SE for n = 129 myocytes (three mice) from the control group and n = 165 myocytes (three mice) from the diabetic group. *P < 0.05.
Indo 1 calcium transients obtained from isolated cardiac myocytes. A: An example of the transients obtained from control and diabetic mice indicating the smaller transient observed in the diabetic group. Analysis of the decay of the transient from peak calcium values (B) revealed a longer decay time in the diabetic group. Measurement of diastolic (C) and absolute transient height (D) calcium levels indicated that both values were significantly lower in the diabetic group, with values being roughly 50% of those measured in the control group. Data represent means ± SE for n = 129 myocytes (three mice) from the control group and n = 165 myocytes (three mice) from the diabetic group. *P < 0.05.

Measurement of SR calcium leakage in myocytes from control and diabetic mice. A: An example of leakage measurements made in cardiac myocytes. The dashed line represents the baseline indo 1 ratio. B: The percentage of calcium leakage from the SR was observed to be approximately three times higher in the diabetic (n = 30 myocytes) than in the control group (n = 20 myocytes). *P < 0.05.
Measurement of SR calcium leakage in myocytes from control and diabetic mice. A: An example of leakage measurements made in cardiac myocytes. The dashed line represents the baseline indo 1 ratio. B: The percentage of calcium leakage from the SR was observed to be approximately three times higher in the diabetic (n = 30 myocytes) than in the control group (n = 20 myocytes). *P < 0.05.
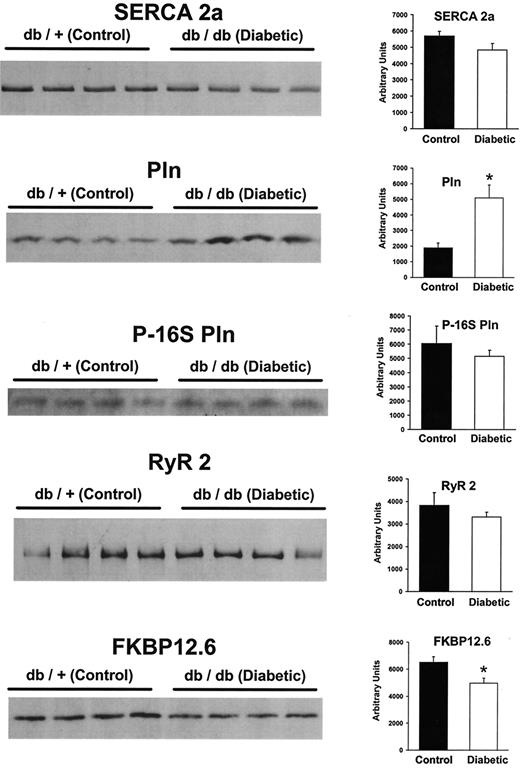
Western blots of various SR proteins obtained from control and diabetic hearts. SERCA2a protein content was slightly (NS) decreased in the diabetic group (average densitometric scan values shown to the right of the blot). In contrast, phospholamban (Pln) levels were two to three times higher in the diabetic group. Results of the analysis of the phosphorylation status of serine residue 16 of Pln (P-16S Pln) were not different between the groups. The expression of the ryanodine receptor (RyR2) did not differ between the two groups; however, a significant decrease in FKBP 12.6 was observed. All lanes were loaded with 20 μg protein, with the exception of the RyR2 blot, where lanes were loaded with 50 μg protein. *P < 0.05.
Western blots of various SR proteins obtained from control and diabetic hearts. SERCA2a protein content was slightly (NS) decreased in the diabetic group (average densitometric scan values shown to the right of the blot). In contrast, phospholamban (Pln) levels were two to three times higher in the diabetic group. Results of the analysis of the phosphorylation status of serine residue 16 of Pln (P-16S Pln) were not different between the groups. The expression of the ryanodine receptor (RyR2) did not differ between the two groups; however, a significant decrease in FKBP 12.6 was observed. All lanes were loaded with 20 μg protein, with the exception of the RyR2 blot, where lanes were loaded with 50 μg protein. *P < 0.05.
Mouse body weight and plasma glucose levels
| Body wt (g) | Plasma glucose (mg/dl) | |
|---|---|---|
| db/+ control | 29.4 ± 0.7 | 216 ± 10 |
| db/db diabetic | 48.1 ± 0.7* | 489 ± 44* |
| Body wt (g) | Plasma glucose (mg/dl) | |
|---|---|---|
| db/+ control | 29.4 ± 0.7 | 216 ± 10 |
| db/db diabetic | 48.1 ± 0.7* | 489 ± 44* |
Data are means ± SE from n = 9 mice.
P < 0.05 vs. control group.
Article Information
This study was supported by National Institutes of Health Grants HL-66917 and HL-52946.