Ophthalmology
Abstract
Neovascular age-related macular degeneration (nAMD) remains a major cause of visual impairment and puts considerable burden on patients and health care systems. L-DOPA-treated Parkinson Disease (PD) patients have been shown to be partially protected from nAMD, but the mechanism remains unknown. Using murine models, combining 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD and laser-induced nAMD, standard PD treatment of L-DOPA/DOPA-decarboxylase inhibitor, or specific dopamine receptor inhibitors, we here demonstrate that L-DOPA treatment-induced increase of dopamine mediated dopamine receptor D2 (DRD2) signaling inhibits choroidal neovascularization independently of MPTP-associated nigrostriatal pathway lesion. Analyzing a retrospective cohort of more than two hundred thousand nAMD patients receiving anti-VEGF treatment from the French nationwide insurance database, we show that DRD2-agonist treated (PD) patients have a significantly delayed age of onset for nAMD (81.4 (±7.0) vs 79.4 (±8.1) years old, respectively, p<0.0001) and reduced need for anti-VEGF therapies (-0.6 injections per 100 mg/day daily dose of DRD2 agonists the second year of treatment), similar to the L-DOPA treatment. While providing a mechanistic explanation for an intriguing epidemiological observation, our findings suggest that systemic DRD2 agonists might constitute an adjuvant therapy to delay and reduce the need for anti-VEGF therapy in nAMD patients.
Authors
Thibaud Mathis, Florian Baudin, Anne-Sophie Mariet, Sébastien Augustin, Marion Bricout, Lauriane Przegralek, Christophe Roubeix, Éric Benzenine, Guillaume Blot, Caroline Nous, Laurent Kodjikian, Martine Mauget-Faÿsse, José-Alain Sahel, Robin Plevin, Christina Zeitz, Cécile Delarasse, Xavier Guillonneau, Catherine Creuzot-Garcher, Catherine Quantin, Stéphane Hunot, Florian Sennlaub
Abstract
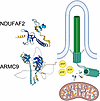
Mitochondria-related neurodegenerative diseases have been implicated in the disruption of primary cilia function. Mutation in an intrinsic mitochondrial complex I component NDUFAF2 has been identified in Leigh syndrome, a severe inherited mitochondriopathy. Mutations in ARMC9, which encodes a basal body protein, cause Joubert syndrome, a ciliopathy with defects in the brain, kidney, and eye. Here, we report a mechanistic link between mitochondria metabolism and primary cilia signaling. We discovered that loss of NDUFAF2 caused both mitochondrial and ciliary defects in vitro and in vivo and identified NDUFAF2 as a binding partner for ARMC9. We also found that NDUFAF2 was both necessary and sufficient for cilia formation and that exogenous expression of NDUFAF2 rescued the ciliary and mitochondrial defects observed in cells from patients with known ARMC9 deficiency. NAD+ supplementation restored mitochondrial and ciliary dysfunction in ARMC9-deficient cells and zebrafish and ameliorated the ocular motility and motor deficits of a patient with ARMC9 deficiency. The present results provide a compelling mechanistic link, supported by evidence from human studies, between primary cilia and mitochondrial signaling. Importantly, our findings have significant implications for the development of therapeutic approaches targeting ciliopathies.
Authors
Chien-Hui Lo, Zhiquan Liu, Siyu Chen, Frank Lin, Andrew R. Berneshawi, Charles Q. Yu, Euna B. Koo, Tia J. Kowal, Ke Ning, Yang Hu, Won-Jing Wang, Y. Joyce Liao, Yang Sun
Abstract
Background Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations (RVCL-S) is a rare, autosomal dominant, universally fatal disease without effective treatment options. This study explores the safety and preliminary efficacy of crizanlizumab, a humanized monoclonal antibody against P-selectin approved for the prevention of sickle cell crises, in slowing retinal nonperfusion and preserving vision in patients with RVCL-S.METHODS Eleven patients with RVCL-S with confirmed exonuclease 3 prime repair exonuclease 1 (TREX1) mutations received monthly crizanlizumab infusions over 2 years. The study measured the nonperfusion index within 3 retinal zones and the total retina with fluorescein angiography, visual acuity, intraocular pressure (IOP), and optical coherence tomography central subfield thickness (CST) at baseline, 1 year, and 2 years. A mixed repeated-measures analysis was performed to assess the progression rates and changes from baseline.RESULTS Eleven participants received crizanlizumab infusions. All of the participants tolerated crizanlizumab well, with 8 of 11 (72.7%) reporting mild adverse effects such as nausea, fatigue, and gastrointestinal symptoms. The change in total retinal nonperfusion was 7.22% [4.47, 9.97] in year 1 and –0.69% [–4.06, 2.68] in year 2 (P < 0.001). In the mid periphery, the change in nonperfusion was 10.6% [5.1, 16.1] in year 1 and –0.68% [–3.98, 5.35] in year 2 (P < 0.01), demonstrating a reduction in progression of nonperfusion in the second year of treatment. Visual acuity, IOP, and CST remained stable.CONCLUSION Crizanlizumab has an acceptable safety profile. These results show promising potential for examining crizanlizumab in larger studies of RVCL-S and similar small-vessel diseases and for using the retina as a biomarker for systemic disease.Trial registration ClinicalTrials.gov NCT04611880.FUNDING The Clayco Foundation; DeNardo Education and Research Foundation Grant; Jeffrey T. Fort Innovation Fund; Siteman Retina Research Fund; unrestricted grant from Research to Prevent Blindness Inc.; National Heart,Lung, and Blood Institute (NHLBI), NIH (R01HL129241); National Institute of Neurological Disorders and Stroke (NINDS), NIH (RF1NS116565).
Authors
Wilson X. Wang, Dan Spiegelman, P. Kumar Rao, Andria L. Ford, Rajendra S. Apte
Abstract
While dysfunction and death of light-detecting photoreceptor cells underlie most inherited retinal dystrophies, knowledge of the species-specific details of human rod and cone photoreceptor cell development remains limited. Here, we generate retinal organoids carrying retinal disease-causing variants in NR2E3, as well as isogenic and unrelated controls. Organoids were sampled using single-cell RNA sequencing across the developmental window encompassing photoreceptor specification, emergence, and maturation. Using scRNAseq data, we reconstruct the rod photoreceptor developmental lineage and identify a branchpoint unique to the disease state. We show that the rod-specific transcription factor NR2E3 is required for the proper expression of genes involved in phototransduction, including rhodopsin, which is absent in divergent rods. NR2E3-null rods additionally misexpress several cone-specific phototransduction genes. Using joint multimodal single-cell sequencing, we further identify putative regulatory sites where rod-specific factors act to steer photoreceptor cell development. Finally, we show that rod-committed photoreceptor cells form and persist throughout life in a patient with NR2E3-associated disease. Importantly, these findings are strikingly different than those observed in Nr2e3 rodent models. Together, these data provide a roadmap of human photoreceptor development and leverage patient iPSCs to define the specific roles of rod transcription factors in photoreceptor cell emergence and maturation in health and disease.
Authors
Nathaniel K. Mullin, Laura R. Bohrer, Andrew P. Voigt, Lola P. Lozano, Allison T. Wright, Vera L. Bonilha, Robert F. Mullins, Edwin M. Stone, Budd A. Tucker
Abstract
Current treatments for neurodegenerative diseases and neural injuries face major challenges, primarily due to the diminished regenerative capacity of neurons in the mammalian central nervous system (CNS) as they mature. Here, we investigated the role of Ezh2, a histone methyltransferase, in regulating mammalian axon regeneration. We found that Ezh2 declined in the mouse nervous system during maturation but was upregulated in adult dorsal root ganglion neurons following peripheral nerve injury to facilitate spontaneous axon regeneration. In addition, overexpression of Ezh2 in retinal ganglion cells in the CNS promoted optic nerve regeneration via both histone methylation-dependent and -independent mechanisms. Further investigation revealed that Ezh2 fostered axon regeneration by orchestrating the transcriptional silencing of genes governing synaptic function and those inhibiting axon regeneration, while concurrently activating various factors that support axon regeneration. Notably, we demonstrated that GABA transporter 2 encoded by Slc6a13 acted downstream of Ezh2 to control axon regeneration. Overall, our study underscores the potential of modulating chromatin accessibility as a promising strategy for promoting CNS axon regeneration.
Authors
Xue-Wei Wang, Shu-Guang Yang, Ming-Wen Hu, Rui-Ying Wang, Chi Zhang, Anish R. Kosanam, Arinze J. Ochuba, Jing-Jing Jiang, Ximei Luo, Yun Guan, Jiang Qian, Chang-Mei Liu, Feng-Quan Zhou

Abstract
Type 2 diabetes mellitus (T2DM), characterized by hyperglycemia and dyslipidemia, leads to nonproliferative diabetic retinopathy (NPDR). NPDR is associated with blood-retina barrier disruption, plasma exudates, microvascular degeneration, elevated inflammatory cytokine levels, and monocyte (Mo) infiltration. Whether and how the diabetes-associated changes in plasma lipid and carbohydrate levels modify Mo differentiation remains unknown. Here, we show that mononuclear phagocytes (MPs) in areas of vascular leakage in DR donor retinas expressed perilipin 2 (PLIN2), a marker of intracellular lipid load. Strong upregulation of PLIN2 was also observed when healthy donor Mos were treated with plasma from patients with T2DM or with palmitate concentrations typical of those found in T2DM plasma, but not under high-glucose conditions. PLIN2 expression correlated with the expression of other key genes involved in lipid metabolism (ACADVL, PDK4) and the DR biomarkers ANGPTL4 and CXCL8. Mechanistically, we show that lipid-exposed MPs induced capillary degeneration in ex vivo explants that was inhibited by pharmaceutical inhibition of PPARγ signaling. Our study reveals a mechanism linking dyslipidemia-induced MP polarization to the increased inflammatory cytokine levels and microvascular degeneration that characterize NPDR. This study provides comprehensive insights into the glycemia-independent activation of Mos in T2DM and identifies MP PPARγ as a target for inhibition of lipid-activated MPs in DR.
Authors
Guillaume Blot, Rémi Karadayi, Lauriane Przegralek, Thérèse-Marie Sartoris, Hugo Charles-Messance, Sébastien Augustin, Pierre Negrier, Frédéric Blond, Frida Paulina Muñiz-Ruvalcaba, David Rivera-de la Parra, Lucile Vignaud, Aude Couturier, José-Alain Sahel, Niyazi Acar, Aida Jimenez-Corona, Cécile Delarasse, Yonathan Garfias, Florian Sennlaub, Xavier Guillonneau
Abstract
Clinical genome editing is emerging for rare disease treatment, but one of the major limitations is the targeting of CRISPR editors delivery. We delivered base editors to the retinal pigmented epithelium (RPE) in the mouse eye using silica nanocapsules (SNC) as a treatment for retinal degeneration. Leber Congenital Amaurosis (LCA16) is a rare pediatric blindness caused by point mutations in the KCNJ13 gene, a loss-of-function inwardly rectifying potassium channel (Kir7.1) in the RPE. SNC carrying adenine base editor (ABE8e) mRNA and single-guide RNA precisely and efficiently corrected KCNJ13W53X/W53X mutation. Editing in both patient fibroblasts (47%) and human-induced pluripotent stem cell-derived RPE (LCA16-iPSC-RPE) (17%) had a negligible off-target response. We detected functional Kir7.1 channels in the edited LCA16-iPSC-RPE. In the LCA16 mouse model (Kcnj13W53X/+∆R), RPE cells targeted SNC delivery of ABE8e mRNA preserved normal visual function measured by full-field electroretinogram (ERG). Moreover, multifocal ERG confirmed the topographic measure of electrical activity primarily originating from the edited retinal area at the injection site. Preserved retina structure, post-treatment, was established by Optical Coherence Tomography (OCT). This preclinical validation of targeted ion channel functional rescue, a challenge for pharmacological and genomic interventions, reinforced the effectiveness of nonviral genome editing therapy for rare inherited disorders.
Authors
Meha Kabra, Pawan K. Shahi, Yuyuan Wang, Divya Sinha, Allison Spillane, Gregory A. Newby, Shivani Saxena, Yao Tong, Yu Chang, Amr A Abdeen, Kimberly L. Edwards, Cole O. Theisen, David R. Liu, David M. Gamm, Shaoqin Gong, Krishanu Saha, Bikash R. Pattnaik
Abstract
Many patients with diabetic eye disease respond inadequately to anti-VEGF therapies, implicating additional vasoactive mediators in its pathogenesis. We demonstrate that levels of angiogenic proteins regulated by hypoxia-inducible factor (HIF)-1 and -2 (HIFs) remain elevated in diabetic eyes despite treatment with anti-VEGF therapy. Conversely, by inhibiting HIFs we normalized the expression of multiple vasoactive mediators in mouse models of diabetic eye disease. Accumulation of HIFs and HIF-regulated vasoactive mediators in hyperglycemic animals was observed in the absence of tissue hypoxia, suggesting that targeting HIFs may be an effective early treatment for diabetic retinopathy. However, while the HIF-inhibitor acriflavine prevented retinal vascular hyperpermeability in diabetic mice for several months following a single intraocular injection, accumulation of acriflavine in the retina resulted in retinal toxicity over time, raising concerns for its use in patients. Conversely, 32-134D, a recently developed HIF inhibitor structurally unrelated to acriflavine, was not toxic to the retina, yet effectively inhibited HIF accumulation and normalized HIF-regulated gene expression in mice and in human retinal organoids. Intraocular administration of 32-134D prevented retinal neovascularization and vascular hyperpermeability in mice. These results provide the foundation for clinical studies assessing 32-134D for the treatment of patients with diabetic eye disease.
Authors
Jing Zhang, Deepti Sharma, Aumreetam Dinabandhu, Jaron Sanchez, Brooks Applewhite, Kathleen Jee, Monika Deshpande, Miguel Flores-Bellver, Ming-Wen Hu, Chuanyu Guo, Shaima Salman, Yousang Hwang, Nicole M. Anders, Michelle A. Rudek, Jiang Qian, Valeria Canto-Soler, Gregg L. Semenza, Silvia Montaner, Akrit Sodhi

Abstract
Patient-derived induced pluripotent stem cells (iPSCs) provide a powerful tool for identifying cellular and molecular mechanisms of disease. Macular telangiectasia type 2 (MacTel) is a rare, late-onset degenerative retinal disease with an extremely heterogeneous genetic architecture, lending itself to the use of iPSCs. Whole-exome sequencing screens and pedigree analyses have identified rare causative mutations that account for less than 5% of cases. Metabolomic surveys of patient populations and GWAS have linked MacTel to decreased circulating levels of serine and elevated levels of neurotoxic 1-deoxysphingolipids (1-dSLs). However, retina-specific, disease-contributing factors have yet to be identified. Here, we used iPSC-differentiated retinal pigmented epithelial (iRPE) cells derived from donors with or without MacTel to screen for novel cell-intrinsic pathological mechanisms. We show that MacTel iRPE cells mimicked the low serine levels observed in serum from patients with MacTel. Through RNA-Seq and gene set enrichment pathway analysis, we determined that MacTel iRPE cells are enriched in cellular stress pathways and dysregulation of central carbon metabolism. Using respirometry and mitochondrial stress testing, we functionally validated that MacTel iRPE cells had a reduction in mitochondrial function that was independent of defects in serine biosynthesis and 1-dSL accumulation. Thus, we identified phenotypes that may constitute alternative disease mechanisms beyond the known serine/sphingolipid pathway.
Authors
Kevin T. Eade, Brendan Robert E. Ansell, Sarah Giles, Regis Fallon, Sarah Harkins-Perry, Takayuki Nagasaki, Simone Tzaridis, Martina Wallace, Elizabeth A. Mills, Samaneh Farashi, Alec Johnson, Lydia Sauer, Barbara Hart, Elena D. Rubio, Melanie Bahlo, Christian Metallo, Rando Allikmets, Marin L. Gantner, Paul S. Bernstein, Martin Friedlander
Abstract
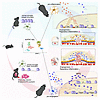
Pathological neovascularization in age-related macular degeneration (nvAMD) drives the principal cause of blindness in the elderly. While there is a robust genetic association between genes of innate immunity and AMD, genome-to-phenome relationships are low, suggesting a critical contribution of environmental triggers of disease. Possible insight comes from the observation that a past history of infection with pathogens such as Chlamydia pneumoniae, or other systemic inflammation, can predispose to nvAMD in later life. Using a mouse model of nvAMD with prior C. pneumoniae infection, endotoxin exposure, and genetic ablation of distinct immune cell populations, we demonstrated that peripheral infections elicited epigenetic reprogramming that led to a persistent memory state in retinal CX3CR1+ mononuclear phagocytes (MNPs). The immune imprinting persisted long after the initial inflammation had subsided and ultimately exacerbated choroidal neovascularization in a model of nvAMD. Single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) identified activating transcription factor 3 (ATF3) as a central mediator of retina-resident MNP reprogramming following peripheral inflammation. ATF3 polarized MNPs toward a reparative phenotype biased toward production of proangiogenic factors in response to subsequent injury. Therefore, a past history of bacterial endotoxin–induced inflammation can lead to immunological reprograming within CNS-resident MNPs and aggravate pathological angiogenesis in the aging retina.
Authors
Masayuki Hata, Maki Hata, Elisabeth M.M.A. Andriessen, Rachel Juneau, Frédérique Pilon, Sergio Crespo-Garcia, Roberto Diaz-Marin, Vera Guber, Francois Binet, Frédérik Fournier, Manuel Buscarlet, Caroline Grou, Virginie Calderon, Emilie Heckel, Heather J. Melichar, Jean-Sebastien Joyal, Ariel M. Wilson, Przemyslaw Sapieha




Copyright © 2024 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)