Issue published July 1, 2024 Previous issue

- Volume 134, Issue 13
Go to section:
On the cover: KLF15 deficiency promotes white-to-beige adipocyte conversion
Li et al. report that KLF15 is required for maintenance of white adipocyte properties in subcutaneous fat depots and is a potential therapeutic target for obesity and associated diseases. The cover image shows multilocular beige adipocytes in a subcutaneous depot of a Klf15-deficient mouse. Image credit: Liang Li.
-
100th Anniversary Viewpoints
×
Abstract
Authors
Mary E. Klotman, Barton F. Haynes

-
Review Series
×
Abstract
Cancer risk is modulated by hereditary and somatic mutations, exposures, age, sex, and gender. The mechanisms by which sex and gender work alone and in combination with other cancer risk factors remain underexplored. In general, cancers that occur in both the male and female sexes occur more commonly in XY compared with XX individuals, regardless of genetic ancestry, geographic location, and age. Moreover, XY individuals are less frequently cured of their cancers, highlighting the need for a greater understanding of sex and gender effects in oncology. This will be necessary for optimal laboratory and clinical cancer investigations. To that end, we review the epigenetics of sexual differentiation and its effect on cancer hallmark pathways throughout life. Specifically, we will touch on how sex differences in metabolism, immunity, pluripotency, and tumor suppressor functions are patterned through the epigenetic effects of imprinting, sex chromosome complement, X inactivation, genes escaping X inactivation, sex hormones, and life history.
Authors
Joshua B. Rubin, Tamara Abou-Antoun, Joseph E. Ippolito, Lorida Llaci, Camryn T. Marquez, Jason P. Wong, Lihua Yang
×Abstract
Biological sex is an important modifier of physiology and influences pathobiology in many diseases. While heart disease is the number one cause of death worldwide in both men and women, sex differences exist at the organ and cellular scales, affecting clinical presentation, diagnosis, and treatment. In this Review, we highlight baseline sex differences in cardiac structure, function, and cellular signaling and discuss the contribution of sex hormones and chromosomes to these characteristics. The heart is a remarkably plastic organ and rapidly responds to physiological and pathological cues by modifying form and function. The nature and extent of cardiac remodeling in response to these stimuli are often dependent on biological sex. We discuss organ- and molecular-level sex differences in adaptive physiological remodeling and pathological cardiac remodeling from pressure and volume overload, ischemia, and genetic heart disease. Finally, we offer a perspective on key future directions for research into cardiac sex differences.
Authors
Thomas G. Martin, Leslie A. Leinwand

-
Commentaries
×
Abstract
Mechanical stress from cardiomyocyte contraction causes misfolded sarcomeric protein replacement. Sarcomeric maintenance utilizes localized pools of mRNAs and translation machinery, yet the importance of localized translation remains unclear. In this issue of the JCI, Haddad et al. identify the Z-line as a critical site for localized translation of sarcomeric proteins, mediated by ribosomal protein SA (RPSA). RPSA localized ribosomes at Z-lines and was trafficked via microtubules. Cardiomyocyte-specific loss of RPSA in mice resulted in mislocalized protein translation and caused structural dilation from myocyte atrophy. These findings demonstrate the necessity of RPSA-dependent spatially localized translation for sarcomere maintenance and cardiac structure and function.
Authors
Abigail Nagle, Michael Regnier, Jennifer Davis
×Abstract
Multiple approaches have targeted voltage-gated sodium (Nav) channels for analgesia. In this issue of the JCI, Shin et al. identified a peptide aptamer, NaViPA1, carrying a short polybasic motif flanked by serine residues in a structurally disordered region of loop 1 in tetrodotoxin-sensitive (TTX-S) but not tetrodotoxin-resistant (TTX-R) channels. NaViPA1h inhibited TTX-S NaV channels and attenuated excitability of sensory neurons. Delivery of NaViPA1 in vivo via adeno-associated virions restricted its expression to peripheral sensory neurons and induced analgesia in rats. Targeting of short linear motifs in this manner may provide a gene therapy modality, with minimal side effects due to its peripherally-restricted biodistribution, which opens up a therapeutic strategy for hyperexcitability disorders, including pain.
Authors
Sulayman D. Dib-Hajj, Stephen G. Waxman
×Abstract
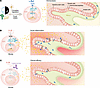
Type 3 innate lymphoid cells (ILC3s) are key regulators of intestinal homeostasis and epithelial barrier integrity. In this issue of the JCI, Cao and colleagues found that a sensor of endoplasmic reticulum (ER) stress, the inositol-requiring kinase 1α/X-box–binding protein 1 (IRE1α/XBP1) pathway, fine-tuned the functions of ILC3s. Activation of IRE1α and XBP1 in ILC3s limited intestinal inflammation in mice and correlated with the efficacy of ustekinumab, an IL-12/IL-23 blocker, in patients with Crohn’s disease. These results advance our understanding in the use of ILCs as biomarkers not only to predict disease outcomes but also to indicate the response to biologicals in patients with inflammatory bowel disease.
Authors
Cinzia Fionda, Giuseppe Sciumè
×Abstract
Cystic fibrosis is a debilitating disease characterized by a poor medical prognosis due to devastating lung injury. Recent medical advances targeting the major genetic mutation ΔF508 of the cystic fibrosis transmembrane conductance regulator (CFTR) protein have dramatically increased the lifespan of patients with this mutation. This development has led to major changes in the field and has pushed research beyond the ion transport nature of cystic fibrosis and toward multiorgan physiological reprogramming. In this issue of the JCI, Bae, Kim, and colleagues utilized a large animal pig model prior to the onset of disease. They revealed metabolic reprogramming and organ crosstalk that occurred prior to disease progression. These findings provide paradigm-shifting insight into this complex disease.
Authors
Sunder Sims-Lucas, Eric S. Goetzman, Thomas R. Kleyman


-
Research Letter
×
Abstract
Authors
Jarmila Stremenova Spegarova, Praisoody Sinnappurajar, Dalila Al Julandani, Rokas Navickas, Helen Griffin, Manisha Ahuja, Angela Grainger, Katie Livingstone, Gillian I. Rice, Fraser Sutherland, Corinne Hayes, Simon Parke, Lewis Pang, Marion R. Roderick, Mary Slatter, Yanick Crow, Athimalaipet V. Ramanan, Sophie Hambleton

-
Research Articles
×
Abstract
Newborn mammalian cardiomyocytes quickly transition from a fetal to an adult phenotype that utilizes mitochondrial oxidative phosphorylation but loses mitotic capacity. We tested whether forced reversal of adult cardiomyocytes back to a fetal glycolytic phenotype would restore proliferative capacity. We deleted Uqcrfs1 (mitochondrial Rieske iron-sulfur protein, RISP) in hearts of adult mice. As RISP protein decreased, heart mitochondrial function declined, and glucose utilization increased. Simultaneously, the hearts underwent hyperplastic remodeling during which cardiomyocyte number doubled without cellular hypertrophy. Cellular energy supply was preserved, AMPK activation was absent, and mTOR activation was evident. In ischemic hearts with RISP deletion, new cardiomyocytes migrated into the infarcted region, suggesting the potential for therapeutic cardiac regeneration. RNA sequencing revealed upregulation of genes associated with cardiac development and proliferation. Metabolomic analysis revealed a decrease in α-ketoglutarate (required for TET-mediated demethylation) and an increase in S-adenosylmethionine (required for methyltransferase activity). Analysis revealed an increase in methylated CpGs near gene transcriptional start sites. Genes that were both differentially expressed and differentially methylated were linked to upregulated cardiac developmental pathways. We conclude that decreased mitochondrial function and increased glucose utilization can restore mitotic capacity in adult cardiomyocytes, resulting in the generation of new heart cells, potentially through the modification of substrates that regulate epigenetic modification of genes required for proliferation.
Authors
Gregory B. Waypa, Kimberly A. Smith, Paul T. Mungai, Vincent J. Dudley, Kathryn A. Helmin, Benjamin D. Singer, Clara Bien Peek, Joseph Bass, Lauren Nelson, Sanjiv J. Shah, Gaston Ofman, J. Andrew Wasserstrom, William A. Muller, Alexander V. Misharin, G.R. Scott Budinger, Hiam Abdala-Valencia, Navdeep S. Chandel, Danijela Dokic, Elizabeth Bartom, Shuang Zhang, Yuki Tatekoshi, Amir Mahmoodzadeh, Hossein Ardehali, Edward B. Thorp, Paul T. Schumacker
×Abstract
The smoothened (Smo) receptor facilitates hedgehog signaling between kidney fibroblasts and tubules during acute kidney injury (AKI). Tubule-derived hedgehog is protective in AKI, but the role of fibroblast-selective Smo is unclear. Here, we report that Smo-specific ablation in fibroblasts reduced tubular cell apoptosis and inflammation, enhanced perivascular mesenchymal cell activities, and preserved kidney function after AKI. Global proteomics of these kidneys identified extracellular matrix proteins, and nidogen-1 glycoprotein in particular, as key response markers to AKI. Intriguingly, Smo was bound to nidogen-1 in cells, suggesting that loss of Smo could affect nidogen-1 accessibility. Phosphoproteomics revealed that the ‘AKI protector’ Wnt signaling pathway was activated in these kidneys. Mechanistically, nidogen-1 interacted with integrin β1 to induce Wnt in tubules to mitigate AKI. Altogether, our results support that fibroblast-selective Smo dictates AKI fate through cell-matrix interactions, including nidogen-1, and offers a robust resource and path to further dissect AKI pathogenesis.
Authors
Yuan Gui, Haiyan Fu, Zachary Palanza, Jianling Tao, Yi-Han Lin, Wenjian Min, Yi Qiao, Christopher Bonin, Geneva Hargis, Yuanyuan Wang, Peng Yang, Donald L. Kreutzer, Yanlin Wang, Yansheng Liu, Yanbao Yu, Youhua Liu, Dong Zhou
×Abstract
One of the features of pathological cardiac hypertrophy is enhanced translation and protein synthesis. Translational inhibition has been shown to be an effective means of treating cardiac hypertrophy, although system-wide side effects are common. Regulators of translation, such as cardiac-specific long noncoding RNAs (lncRNAs), could provide new, more targeted therapeutic approaches to inhibit cardiac hypertrophy. Therefore, we generated mice lacking a previously identified lncRNA named CARDINAL to examine its cardiac function. We demonstrate that CARDINAL is a cardiac-specific, ribosome-associated lncRNA and show that its expression was induced in the heart upon pathological cardiac hypertrophy and that its deletion in mice exacerbated stress-induced cardiac hypertrophy and augmented protein translation. In contrast, overexpression of CARDINAL attenuated cardiac hypertrophy in vivo and in vitro and suppressed hypertrophy-induced protein translation. Mechanistically, CARDINAL interacted with developmentally regulated GTP-binding protein 1 (DRG1) and blocked its interaction with DRG family regulatory protein 1 (DFRP1); as a result, DRG1 was downregulated, thereby modulating the rate of protein translation in the heart in response to stress. This study provides evidence for the therapeutic potential of targeting cardiac-specific lncRNAs to suppress disease-induced translational changes and to treat cardiac hypertrophy and heart failure.
Authors
Xin He, Tiqun Yang, Yao Wei Lu, Gengze Wu, Gang Dai, Qing Ma, Mingming Zhang, Huimin Zhou, Tianxin Long, Youchen Yan, Zhuomin Liang, Chen Liu, William T. Pu, Yugang Dong, Jingsong Ou, Hong Chen, John D. Mably, Jiangui He, Da-Zhi Wang, Zhan-Peng Huang
×Abstract
This study reports that targeting intrinsically disordered regions of the voltage-gated sodium channel 1.7 (NaV1.7) protein facilitates discovery of sodium channel inhibitory peptide aptamers (NaViPA) for adeno-associated virus–mediated (AAV-mediated), sensory neuron–specific analgesia. A multipronged inhibition of INa1.7, INa1.6, INa1.3, and INa1.1 — but not INa1.5 and INa1.8 — was found for a prototype and named NaViPA1, which was derived from the NaV1.7 intracellular loop 1, and is conserved among the TTXs NaV subtypes. NaViPA1 expression in primary sensory neurons (PSNs) of dorsal root ganglia (DRG) produced significant inhibition of TTXs INa but not TTXr INa. DRG injection of AAV6-encoded NaViPA1 significantly attenuated evoked and spontaneous pain behaviors in both male and female rats with neuropathic pain induced by tibial nerve injury (TNI). Whole-cell current clamp of the PSNs showed that NaViPA1 expression normalized PSN excitability in TNI rats, suggesting that NaViPA1 attenuated pain by reversal of injury-induced neuronal hypersensitivity. IHC revealed efficient NaViPA1 expression restricted in PSNs and their central and peripheral terminals, indicating PSN-restricted AAV biodistribution. Inhibition of sodium channels by NaViPA1 was replicated in the human iPSC-derived sensory neurons. These results summate that NaViPA1 is a promising analgesic lead that, combined with AAV-mediated PSN-specific block of multiple TTXs NaVs, has potential as a peripheral nerve–restricted analgesic therapeutic.
Authors
Seung Min Shin, Brandon Itson-Zoske, Fan Fan, Yucheng Xiao, Chensheng Qiu, Theodore R. Cummins, Quinn H. Hogan, Hongwei Yu
×Abstract
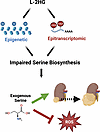
Tumor cells are known to undergo considerable metabolic reprogramming to meet their unique demands and drive tumor growth. At the same time, this reprogramming may come at a cost with resultant metabolic vulnerabilities. The small molecule l-2-hydroxyglutarate (l-2HG) is elevated in the most common histology of renal cancer. Similarly to other oncometabolites, l-2HG has the potential to profoundly impact gene expression. Here, we demonstrate that l-2HG remodels amino acid metabolism in renal cancer cells through combined effects on histone methylation and RNA N6-methyladenosine. The combined effects of l-2HG result in a metabolic liability that renders tumors cells reliant on exogenous serine to support proliferation, redox homeostasis, and tumor growth. In concert with these data, high–l-2HG kidney cancers demonstrate reduced expression of multiple serine biosynthetic enzymes. Collectively, our data indicate that high–l-2HG renal tumors could be specifically targeted by strategies that limit serine availability to tumors.
Authors
Anirban Kundu, Garrett J. Brinkley, Hyeyoung Nam, Suman Karki, Richard Kirkman, Madhuparna Pandit, EunHee Shim, Hayley Widden, Juan Liu, Yasaman Heidarian, Nader H. Mahmoudzadeh, Alexander J. Fitt, Devin Absher, Han-Fei Ding, David K. Crossman, William J. Placzek, Jason W. Locasale, Dinesh Rakheja, Jonathan E. McConathy, Rekha Ramachandran, Sejong Bae, Jason M. Tennessen, Sunil Sudarshan
×Abstract
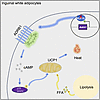
Healthy adipose tissue is essential for normal physiology. There are 2 broad types of adipose tissue depots: brown adipose tissue (BAT), which contains adipocytes poised to burn energy through thermogenesis, and white adipose tissue (WAT), which contains adipocytes that store lipids. However, within those types of adipose, adipocytes possess depot and cell-specific properties that have important implications. For example, the subcutaneous and visceral WAT confers divergent risk for metabolic disease. Further, within a depot, different adipocytes can have distinct properties; subcutaneous WAT can contain adipocytes with either white or brown-like (beige) adipocyte properties. However, the pathways that regulate and maintain this cell and depot-specificity are incompletely understood. Here, we found that the transcription factor KLF15 is required for maintaining white adipocyte properties selectively within the subcutaneous WAT. We revealed that deletion of Klf15 is sufficient to induce beige adipocyte properties and that KLF15’s direct regulation of Adrb1 is a critical molecular mechanism for this process. We uncovered that this activity is cell autonomous but has systemic implications in mouse models and is conserved in primary human adipose cells. Our results elucidate a pathway for depot-specific maintenance of white adipocyte properties that could enable the development of therapies for obesity and associated diseases.
Authors
Liang Li, Brian J. Feldman
×Abstract
Group 3 innate lymphoid cells (ILC3s) are key players in intestinal homeostasis. ER stress is linked to inflammatory bowel disease (IBD). Here, we used cell culture, mouse models, and human specimens to determine whether ER stress in ILC3s affects IBD pathophysiology. We show that mouse intestinal ILC3s exhibited a 24-hour rhythmic expression pattern of the master ER stress response regulator inositol-requiring kinase 1α/X-box–binding protein 1 (IRE1α/XBP1). Proinflammatory cytokine IL-23 selectively stimulated IRE1α/XBP1 in mouse ILC3s through mitochondrial ROS (mtROS). IRE1α/XBP1 was activated in ILC3s from mice exposed to experimental colitis and in inflamed human IBD specimens. Mice with Ire1α deletion in ILC3s (Ire1αΔRorc) showed reduced expression of the ER stress response and cytokine genes including Il22 in ILC3s and were highly vulnerable to infections and colitis. Administration of IL-22 counteracted their colitis susceptibility. In human ILC3s, IRE1 inhibitors suppressed cytokine production, which was upregulated by an IRE1 activator. Moreover, the frequencies of intestinal XBP1s+ ILC3s in patients with Crohn’s disease before administration of ustekinumab, an anti-IL-12/IL-23 antibody, positively correlated with the response to treatment. We demonstrate that a noncanonical mtROS-IRE1α/XBP1 pathway augmented cytokine production by ILC3s and identify XBP1s+ ILC3s as a potential biomarker for predicting the response to anti–IL-23 therapies in IBD.
Authors
Siyan Cao, Jose L. Fachi, Kaiming Ma, Alina Ulezko Antonova, Qianli Wang, Zhangying Cai, Randal J. Kaufman, Matthew A. Ciorba, Parakkal Deepak, Marco Colonna
×Abstract
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene cause cystic fibrosis (CF), a multiorgan disease that is characterized by diverse metabolic defects. However, other than specific CFTR mutations, the factors that influence disease progression and severity remain poorly understood. Aberrant metabolite levels have been reported, but whether CFTR loss itself or secondary abnormalities (infection, inflammation, malnutrition, and various treatments) drive metabolic defects is uncertain. Here, we implemented comprehensive arteriovenous metabolomics in newborn CF pigs, and the results revealed CFTR as a bona fide regulator of metabolism. CFTR loss impaired metabolite exchange across organs, including disruption of lung uptake of fatty acids, yet enhancement of uptake of arachidonic acid, a precursor of proinflammatory cytokines. CFTR loss also impaired kidney reabsorption of amino acids and lactate and abolished renal glucose homeostasis. These and additional unexpected metabolic defects prior to disease manifestations reveal a fundamental role for CFTR in controlling multiorgan metabolism. Such discovery informs a basic understanding of CF, provides a foundation for future investigation, and has implications for developing therapies targeting only a single tissue.
Authors
Hosung Bae, Bo Ram Kim, Sunhee Jung, Johnny Le, Dana van der Heide, Wenjie Yu, Sang Hee Park, Brieanna M. Hilkin, Nicholas D. Gansemer, Linda S. Powers, Taekyung Kang, David K. Meyerholz, Victor L. Schuster, Cholsoon Jang, Michael J. Welsh
×Abstract
Cardiomyocyte sarcomeres contain localized ribosomes, but the factors responsible for their localization and the significance of localized translation are unknown. Using proximity labeling, we identified ribosomal protein SA (RPSA) as a Z-line protein. In cultured cardiomyocytes, the loss of RPSA led to impaired local protein translation and reduced sarcomere integrity. By employing CAS9-expressing mice, along with adeno-associated viruses expressing CRE recombinase and single-guide RNAs targeting Rpsa, we knocked out Rpsa in vivo and observed mislocalization of ribosomes and diminished local translation. These genetic mosaic mice with Rpsa knockout in a subset of cardiomyocytes developed dilated cardiomyopathy, featuring atrophy of RPSA-deficient cardiomyocytes, compensatory hypertrophy of unaffected cardiomyocytes, left ventricular dilation, and impaired contractile function. We demonstrated that RPSA C-terminal domain is sufficient for localization to the Z-lines and that if the microtubule network is disrupted RPSA loses its sarcomeric localization. These findings highlight RPSA as a ribosomal factor essential for ribosome localization to the Z-line, facilitating local translation and sarcomere maintenance.
Authors
Rami Haddad, Omer Sadeh, Tamar Ziv, Itai Erlich, Lilac Haimovich-Caspi, Ariel Shemesh, Jolanda van der Velden, Izhak Kehat
×Abstract
Diffuse midline glioma (DMG) H3K27-altered is one of the most malignant childhood cancers. Radiation therapy remains the only effective treatment yet provides a 5-year survival rate of only 1%. Several clinical trials have attempted to enhance radiation antitumor activity using radiosensitizing agents, although none have been successful. Given this, there is a critical need for identifying effective therapeutics to enhance radiation sensitivity for the treatment of DMG. Using high-throughput radiosensitivity screening, we identified bromo- and extraterminal domain (BET) protein inhibitors as potent radiosensitizers in DMG cells. Genetic and pharmacologic inhibition of BET bromodomain activity reduced DMG cell proliferation and enhanced radiation-induced DNA damage by inhibiting DNA repair pathways. RNA-Seq and the CUT&RUN (cleavage under targets and release using nuclease) analysis showed that BET bromodomain inhibitors regulated the expression of DNA repair genes mediated by H3K27 acetylation at enhancers. BET bromodomain inhibitors enhanced DMG radiation response in patient-derived xenografts as well as genetically engineered mouse models. Together, our results highlight BET bromodomain inhibitors as potential radiosensitizer and provide a rationale for developing combination therapy with radiation for the treatment of DMG.
Authors
Jun Watanabe, Matthew R. Clutter, Michael J. Gullette, Takahiro Sasaki, Eita Uchida, Savneet Kaur, Yan Mo, Kouki Abe, Yukitomo Ishi, Nozomu Takata, Manabu Natsumeda, Samantha Gadd, Zhiguo Zhang, Oren J. Becher, Rintaro Hashizume
×Abstract
Endothelial cells (ECs) in the descending aorta are exposed to high laminar shear stress, and this supports an antiinflammatory phenotype. High laminar shear stress also induces flow-aligned cell elongation and front-rear polarity, but whether these are required for the antiinflammatory phenotype is unclear. Here, we showed that caveolin-1–rich microdomains polarize to the downstream end of ECs that are exposed to continuous high laminar flow. These microdomains were characterized by high membrane rigidity, filamentous actin (F-actin), and raft-associated lipids. Transient receptor potential vanilloid (TRPV4) ion channels were ubiquitously expressed on the plasma membrane but mediated localized Ca2+ entry only at these microdomains where they physically interacted with clustered caveolin-1. These focal Ca2+ bursts activated endothelial nitric oxide synthase within the confines of these domains. Importantly, we found that signaling at these domains required both cell body elongation and sustained flow. Finally, TRPV4 signaling at these domains was necessary and sufficient to suppress inflammatory gene expression and exogenous activation of TRPV4 channels ameliorated the inflammatory response to stimuli both in vitro and in vivo. Our work revealed a polarized mechanosensitive signaling hub in arterial ECs that dampened inflammatory gene expression and promoted cell resilience.
Authors
Soon-Gook Hong, Julianne W. Ashby, John P. Kennelly, Meigan Wu, Michelle Steel, Eesha Chattopadhyay, Rob Foreman, Peter Tontonoz, Elizabeth J. Tarling, Patric Turowski, Marcus Gallagher-Jones, Julia J. Mack
×Abstract
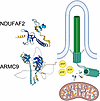
Mitochondria-related neurodegenerative diseases have been implicated in the disruption of primary cilia function. Mutation in an intrinsic mitochondrial complex I component NDUFAF2 has been identified in Leigh syndrome, a severe inherited mitochondriopathy. Mutations in ARMC9, which encodes a basal body protein, cause Joubert syndrome, a ciliopathy with defects in the brain, kidney, and eye. Here, we report a mechanistic link between mitochondria metabolism and primary cilia signaling. We discovered that loss of NDUFAF2 caused both mitochondrial and ciliary defects in vitro and in vivo and identified NDUFAF2 as a binding partner for ARMC9. We also found that NDUFAF2 was both necessary and sufficient for cilia formation and that exogenous expression of NDUFAF2 rescued the ciliary and mitochondrial defects observed in cells from patients with known ARMC9 deficiency. NAD+ supplementation restored mitochondrial and ciliary dysfunction in ARMC9-deficient cells and zebrafish and ameliorated the ocular motility and motor deficits of a patient with ARMC9 deficiency. The present results provide a compelling mechanistic link, supported by evidence from human studies, between primary cilia and mitochondrial signaling. Importantly, our findings have significant implications for the development of therapeutic approaches targeting ciliopathies.
Authors
Chien-Hui Lo, Zhiquan Liu, Siyu Chen, Frank Lin, Andrew R. Berneshawi, Charles Q. Yu, Euna B. Koo, Tia J. Kowal, Ke Ning, Yang Hu, Won-Jing Wang, Y. Joyce Liao, Yang Sun
×Abstract
Impairment of oligodendrocytes and myelin contributes to neurological disorders including multiple sclerosis (MS), stroke, and Alzheimer’s disease. Regeneration of myelin (remyelination) decreases the vulnerability of demyelinated axons, but this repair process commonly fails with disease progression. A contributor to inefficient remyelination is the altered extracellular matrix (ECM) in lesions, which remains to be better defined. We have identified fibulin-2 (FBLN2) as a highly upregulated ECM component in lesions of MS and stroke and in proteome databases of Alzheimer’s disease and traumatic brain injury. Focusing on MS, the inhibitory role of FBLN2 was suggested in the experimental autoimmune encephalomyelitis (EAE) model, in which genetic FBLN2 deficiency improved behavioral recovery by promoting the maturation of oligodendrocytes and enhancing remyelination. Mechanistically, when oligodendrocyte progenitors were cultured in differentiation medium, FBLN2 impeded their maturation into oligodendrocytes by engaging the Notch pathway, leading to cell death. Adeno-associated virus deletion of FBLN2 in astrocytes improved oligodendrocyte numbers and functional recovery in EAE and generated new myelin profiles after lysolecithin-induced demyelination. Collectively, our findings implicate FBLN2 as a hitherto unrecognized injury-elevated ECM, and a therapeutic target, that impairs oligodendrocyte maturation and myelin repair.
Authors
Samira Ghorbani, Cenxiao Li, Brian M. Lozinski, Dorsa Moezzi, Charlotte D’Mello, Yifei Dong, Frank Visser, Hongmin Li, Claudia Silva, Mohammadparsa Khakpour, Colin J. Murray, Marie-Ève Tremblay, Mengzhou Xue, V. Wee Yong
×Abstract
The diversity of structural variants (SVs) in melanoma and how they impact oncogenesis are incompletely known. We performed harmonized analysis of SVs across melanoma histologic and genomic subtypes, and we identified distinct global properties between subtypes. These included the frequency and size of SVs and SV classes, their relation to chromothripsis events, and the impact on cancer-related genes of SVs that alter topologically associated domain (TAD) boundaries. Following our prior identification of double-stranded break repair deficiency in a subset of triple-wild-type cutaneous melanoma, we identified MRE11 and NBN loss-of-function SVs in melanomas with this mutational signature. Experimental knockouts of MRE11 and NBN, followed by olaparib cell viability assays in melanoma cells, indicated that dysregulation of each of these genes may cause sensitivity to PARP inhibitors in cutaneous melanomas. Broadly, harmonized analysis of melanoma SVs revealed distinct global genomic properties and molecular drivers, which may have biological and therapeutic impact.
Authors
Jake R. Conway, Riaz Gillani, Jett Crowdis, Brendan Reardon, Jihye Park, Seunghun Han, Breanna Titchen, Mouadh Benamar, Rizwan Haq, Eliezer M. Van Allen
×Abstract
Ubiquitination plays an essential role in protein stability, subcellular localization, and interactions. Crosstalk between different types of ubiquitination results in distinct biological outcomes for proteins. However, the role of ubiquitination-related crosstalk in lymph node (LN) metastasis and the key regulatory factors controlling this process have not been determined. Using high-throughput sequencing, we found that ubiquitin-conjugating enzyme E2 C (UBE2C) was overexpressed in bladder cancer (BCa) and was strongly associated with an unfavorable prognosis. Overexpression of UBE2C increased BCa lymphangiogenesis and promoted LN metastasis both in vitro and in vivo. Mechanistically, UBE2C mediated sodium-coupled neutral amino acid transporter 2 (SNAT2) monoubiquitination at lysine 59 to inhibit K63-linked polyubiquitination at lysine 33 of SNAT2. Crosstalk between monoubiquitination and K63-linked polyubiquitination increased SNAT2 membrane protein levels by suppressing epsin 1–mediated (EPN1-mediated) endocytosis. SNAT2 facilitated glutamine uptake and metabolism to promote VEGFC secretion, ultimately leading to lymphangiogenesis and LN metastasis in patients with BCa. Importantly, inhibition of UBE2C significantly attenuated BCa lymphangiogenesis in a patient-derived xenograft model. Our results reveal the mechanism by which UBE2C mediates crosstalk between the monoubiquitination and K63-linked polyubiquitination of SNAT2 to promote BCa metastasis and identify UBE2C as a promising target for treating LN-metastatic BCa.
Authors
Wenjie Li, Changhao Chen, Hanhao Zheng, Yan Lin, Mingjie An, Daiyin Liu, Yonghai Zhang, Mingchao Gao, Tianhang Lan, Wang He
×Abstract
Given the global surge in autoimmune diseases, it is critical to evaluate emerging therapeutic interventions. Despite numerous new targeted immunomodulatory therapies, comprehensive approaches to apply and evaluate the effects of these treatments longitudinally are lacking. Here, we leveraged advances in programmable-phage immunoprecipitation methodology to explore the modulation, or lack thereof, of autoantibody profiles, proteome-wide, in both health and disease. Using a custom set of over 730,000 human-derived peptides, we demonstrated that each individual, regardless of disease state, possesses a distinct and complex constellation of autoreactive antibodies. For each individual, the set of resulting autoreactivites constituted a unique immunological fingerprint, or “autoreactome,” that was remarkably stable over years. Using the autoreactome as a primary output, we evaluated the relative effectiveness of various immunomodulatory therapies in altering autoantibody repertoires. We found that therapies targeting B cell maturation antigen (BCMA) profoundly altered an individual’s autoreactome, while anti-CD19 and anti-CD20 therapies had minimal effects. These data both confirm that the autoreactome comprises autoantibodies secreted by plasma cells and strongly suggest that BCMA or other plasma cell–targeting therapies may be highly effective in treating currently refractory autoantibody-mediated diseases.
Authors
Aaron Bodansky, David J.L. Yu, Alysa Rallistan, Muge Kalaycioglu, Jim Boonyaratanakornkit, Damian J. Green, Jordan Gauthier, Cameron J. Turtle, Kelsey Zorn, Brian O’Donovan, Caleigh Mandel-Brehm, James Asaki, Hannah Kortbawi, Andrew F. Kung, Elze Rackaityte, Chung-Yu Wang, Aditi Saxena, Kimberly de Dios, Gianvito Masi, Richard J. Nowak, Kevin C. O’Connor, Hao Li, Valentina E. Diaz, Rowan Saloner, Kaitlin B. Casaletto, Eva Q. Gontrum, Brandon Chan, Joel H. Kramer, Michael R. Wilson, Paul J. Utz, Joshua A. Hill, Shaun W. Jackson, Mark S. Anderson, Joseph L. DeRisi
×Abstract
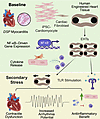
Myocarditis is clinically characterized by chest pain, arrhythmias, and heart failure, and treatment is often supportive. Mutations in DSP, a gene encoding the desmosomal protein desmoplakin, have been increasingly implicated in myocarditis. To model DSP-associated myocarditis and assess the role of innate immunity, we generated engineered heart tissues (EHTs) using human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) from patients with heterozygous DSP truncating variants (DSPtvs) and a gene-edited homozygous deletion cell line (DSP–/–). At baseline, DSP–/– EHTs displayed a transcriptomic signature of innate immune activation, which was mirrored by cytokine release. Importantly, DSP–/– EHTs were hypersensitive to Toll-like receptor (TLR) stimulation, demonstrating more contractile dysfunction compared with isogenic controls. Relative to DSP–/– EHTs, heterozygous DSPtv EHTs had less functional impairment. DSPtv EHTs displayed heightened sensitivity to TLR stimulation, and when subjected to strain, DSPtv EHTs developed functional deficits, indicating reduced contractile reserve compared with healthy controls. Colchicine or NF-κB inhibitors improved strain-induced force deficits in DSPtv EHTs. Genomic correction of DSP p.R1951X using adenine base editing reduced inflammatory biomarker release from EHTs. Thus, EHTs replicate electrical and contractile phenotypes seen in human myocarditis, implicating cytokine release as a key part of the myogenic susceptibility to inflammation. The heightened innate immune activation and sensitivity are targets for clinical intervention.
Authors
Daniel F. Selgrade, Dominic E. Fullenkamp, Ivana A. Chychula, Binjie Li, Lisa Dellefave-Castillo, Adi D. Dubash, Joyce Ohiri, Tanner O. Monroe, Malorie Blancard, Garima Tomar, Cory Holgren, Paul W. Burridge, Alfred L. George Jr., Alexis R. Demonbreun, Megan J. Puckelwartz, Sharon A. George, Igor R. Efimov, Kathleen J. Green, Elizabeth M. McNally
×Abstract
Steatotic donor livers are becoming more and more common in liver transplantation. However, the current use of steatotic grafts is less acceptable than normal grafts due to their higher susceptibility to ischemia/reperfusion (I/R) injury. To investigate the mechanism underlying the susceptibility of steatotic liver to I/R injury, we detected cell death markers and inflammation in clinical donor livers and animal models. We found that caspase-8–mediated hepatic apoptosis is activated in steatotic liver I/R injury. However, ablation of caspase-8 only slightly mitigated steatotic liver I/R injury without affecting inflammation. We further demonstrated that RIPK1 kinase induces both caspase-8–mediated apoptosis and cell death–independent inflammation. Inhibition of RIPK1 kinase significantly protects against steatotic liver I/R injury by alleviating both hepatic apoptosis and inflammation. Additionally, we found that RIPK1 activation is induced by Z-DNA binding protein 1 (ZBP1) but not the canonical TNF-α pathway during steatotic liver I/R injury. Deletion of ZBP1 substantially decreases the steatotic liver I/R injury. Mechanistically, ZBP1 is amplified by palmitic acid–activated JNK pathway in steatotic livers. Upon I/R injury, excessive reactive oxygen species trigger ZBP1 activation by inducing its aggregation independent of the Z-nucleic acids sensing action in steatotic livers, leading to the kinase activation of RIPK1 and the subsequent aggravation of liver injury. Thus, ZBP1-mediated RIPK1-driven apoptosis and inflammation exacerbate steatotic liver I/R injury, which could be targeted to protect steatotic donor livers during transplantation.
Authors
Ran Liu, Huan Cao, Shuhua Zhang, Mao Cai, Tianhao Zou, Guoliang Wang, Di Zhang, Xueling Wang, Jianjun Xu, Shenghe Deng, Tongxi Li, Daichao Xu, Jinyang Gu










In-Press Preview - More
Abstract
Authors
Amandeep Jutla, Lauren C. Shuffrey, Stephen J. Guter, Kally C. O'Reilly, George M. Anderson, James S. Sutcliffe, Edwin H. Cook, Jeremy Veenstra-VanderWeele
Abstract
Cell cycle regulation is largely abnormal in cancers. Molecular understanding and therapeutic targeting of the aberrant cell cycle are essentially meaningful. Here, we identified an under-appreciated Serine/Threonine kinase, CDKL3 (Cyclin-dependent kinase like 3), crucially drives the rapid cell cycle progression and cell growth in cancers. Mechanism-wise, CDKL3 localizes in the nucleus and associates with specific cyclin to directly phosphorylate Retinoblastoma (Rb) for quiescence exit. In parallel, CDKL3 prevents the ubiquitin-proteasomal degradation of CDK4 by direct phosphorylation on T172 to sustain G1 phase advancement. The crucial function of CDKL3 in cancers was demonstrated both in vitro and in vivo. We also designed, synthesized and characterized a first-in-class CDKL3-specific inhibitor, HZ1. HZ1 exhibits greater potency than CDK4/6 (Cyclin-dependent kinase 4/6) inhibitor in pan-cancer treatment by causing cell cycle arrest and overcomes the acquired resistance of the latter. In particular, CDKL3 has significant clinical relevance in colon cancer, and the effectiveness of HZ1 was demonstrated by murine and patient-derived cancer models. Collectively, this work presented an integrated paradigm of cancer cell cycle regulation and suggested CDKL3-targeting as a feasible approach in cancer treatment.
Authors
Haijiao Zhang, Jiahui Lin, Shaoqin Zheng, Lanjing Ma, Zhongqiu Pang, Hongyi Yin, Chengcheng Meng, Yinuo Wang, Qing Han, Xi Zhang, Zexu Li, Liu Cao, Lijun Liu, Teng Fei, Daming Gao, Liang Yang, Xueqiang Peng, Chen Ding, Shixue Wang, Ren Sheng
Abstract
Background: Antibiotic-Refractory Lyme Arthritis (ARLA) involves a complex interplay of T cell responses targeting Borrelia burgdorferi antigens succeeding towards autoantigens by epitope spreading. However, the precise molecular mechanisms driving the pathogenic T cell response in ARLA remain unclear. Our aim was to elucidate the molecular program of disease-specific Th cells. Methods: Using flow cytometry, high-throughput T cell receptor (TCR) sequencing and scRNA-seq of CD4+ Th cells isolated from the joints of European ARLA patients, we aimed at inferring antigen specificity through unbiased analysis of TCR repertoire patterns, identifying surrogate markers for disease-specific TCRs and connecting TCR specificity to transcriptional patterns. Results: PD-1hiHLA-DR+CD4+ effector T cells were clonally expanded within the inflamed joints and persisted throughout disease course. Among these cells, we identified a distinct TCRβ motif restricted to HLA-DRB1*11 or *13 alleles. These alleles, being underrepresented in North American ARLA patients, were unexpectedly prevalent in our European cohort. The identified TCRβ motif served as surrogate marker for a convergent TCR response specific to ARLA, distinguishing it from other rheumatic diseases. In the scRNA-seq dataset, the TCRβ motif particularly mapped to peripheral T helper (TPH) cells displaying signs of sustained proliferation, continuous TCR signaling, and expressing CXCL13 and IFN-γ. Conclusion: By inferring disease-specific TCRs from synovial T cells we identified a convergent TCR response in the joints of ARLA patients that continuously fueled the expansion of TPH cells expressing a pathogenic cytokine effector program. The identified TCRs will aid in uncovering the major antigen targets of the maladaptive immune response. Funding: Supported by the German Research Foundation (DFG) MO 2160/4-1; the Federal Ministry of Education and Research (BMBF; Advanced Clinician Scientist-Program INTERACT; 01EO2108) embedded in the Interdisciplinary Center for Clinical Research (IZKF) of the University Hospital Würzburg; the German Center for Infection Research (DZIF; Clinical Leave Program; TI07.001_007) and the Interdisciplinary Center for Clinical Research (IZKF) Würzburg (Clinician Scientist Program, Z-2/CSP-30).
Authors
Johannes Dirks, Jonas Fischer, Julia Klaussner, Christine Hofmann, Annette Holl-Wieden, Viktoria Buck, Christian Klemann, Hermann J. Girschick, Ignazio Caruana, Florian Erhard, Henner Morbach
Abstract
Cytomegalovirus (CMV) is one of the most common and relevant opportunistic pathogens in immunocompromised individuals such as kidney transplant recipients (KTRs). The exact mechanisms underlying the disability of cytotoxic T cells to provide sufficient protection against CMV in immunosuppressed individuals have not been identified yet. Here, we performed in-depth metabolic profiling of CMV-specific CD8+ T cells in immunocompromised patients and show the development of metabolic dysregulation at the transcriptional, protein, and functional level of CMV-specific CD8+ T cells in KTRs with non-controlled CMV infection. These dysregulations comprise impaired glycolysis and increased mitochondrial stress, which is associated with an intensified expression of the nicotinamide adenine dinucleotide nucleotidase (NADase) CD38. Inhibiting NADase activity of CD38 reinvigorated the metabolism and improved cytokine production of CMV-specific CD8+ T cells. These findings were corroborated in a mouse model of CMV infection under conditions of immunosuppression. Thus, dysregulated metabolic states of CD8+ T cells could be targeted by inhibiting CD38 to reverse hypo-responsiveness in individuals who fail to control chronic viral infection.
Authors
Nils Mülling, Felix M. Behr, Graham A. Heieis, Kristina Boss, Suzanne van Duikeren, Floortje J. van Haften, Iris N. Pardieck, Esmé T.I. van der Gracht, Ward Vleeshouwers, Tetje C. van der Sluis, J. Fréderique de Graaf, Dominique M.B. Veerkamp, Kees L.M.C. Franken, Xin Lei, Lukas van de Sand, Sjoerd H. van der Burg, Marij J.P. Welters, Sebastiaan Heidt, Wesley Huisman, Simon P. Jochems, Martin Giera, Oliver Witzke, Aiko P.J. de Vries, Andreas Kribben, Bart Everts, Benjamin Wilde, Ramon Arens
Abstract
Cystic fibrosis (CF) results from mutations in the CFTR anion channel, ultimately leading to diminished transepithelial anion secretion and mucociliary clearance. CFTR correctors are therapeutics that restore the folding/trafficking of mutated CFTR to the plasma membrane. The BKCa potassium channel is also critical for maintaining lung ASL volume. Here, we show the CFTR corrector, VX-445 (Elexacaftor), a component of Trikafta, induces K+ secretion across WT and F508del CFTR primary human bronchial epithelial cells (HBEs), which was entirely inhibited by the BKCa antagonist paxilline. Similar results were observed with VX-121 – a corrector under clinical evaluation. Whole-cell patch-clamp recordings confirmed potentiated channel activity from CFTR correctors on the BKCa α-subunit, and excised patch-clamp recordings demonstrated a significant increase in open probability. In mesenteric artery, VX-445 induced a paxilline-sensitive vasorelaxation of preconstricted arteries. VX-445 also reduced action potential firing frequency in primary hippocampal and cortical neurons. VX-445 effects were observed at low micomolar concentrations (1-10 µM) – within the range reported in plasma and tissues from CF patients. We raise the possibilities that CFTR correctors gain additional clinical benefit by activation of BKCa in the lung, yet may lead to adverse events through BKCa activation, elsewhere.
Authors
Aaron Kolski-Andreaco, Stefanie Taiclet, Michael M. Myerburg, john sembrat, Robert J. Bridges, Adam C. Straub, Zachary P. Wills, Michael B. Butterworth, Daniel C. Devor
View more articles by topic:
Sign up for email alerts
JCI's 100th anniversary

JCI celebrates a century of publishing scientific discoveries with a special collection highlighting major innovations in medicine and key contributing mechanistic studies.
Review Series - More
Series edited by Barbara Stranger
Sex Differences in Medicine
Series edited by Barbara Stranger
Biological sex profoundly influences disease risk, pathogenesis, progression, and treatment, but there are persistent gaps in the study of sex differences that span all areas of medicine. Reviews in this series will examine sex as a biological variable in cancer, metabolism, cardiovascular disease, autoimmunity, and more and highlight the potential to leverage these sex differences to optimize therapies for all.
×
Author's Take - More



Copyright © 2024 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)