


Abstract
Extracorporeal photochemotherapy (ECP) is a cancer immunotherapy for cutaneous T-cell lymphoma (CTCL) operative in more than 350 centers worldwide. Although its efficacy and favorable safety profile have driven its widespread use, elucidation of its underlying mechanism has been difficult. In this study, we identify the principal contributors to the anticancer immunotherapeutic effects of ECP, with the goal of enhancing potency and broadening applicability to additional malignancies. First, we scaled down the clinical ECP leukocyte-processing device to mouse size. Second, we used that miniaturized device to produce a cellular vaccine that regularly initiated therapeutic antimelanoma immunity. Third, we individually subtracted key factors from either the immunizing inoculum or the treated animal to ascertain their contribution to the in vivo antimelanoma response. Platelet-signaled monocyte-to-dendritic cell (DC) differentiation followed by sorting/processing/presentation of tumor antigens derived from internalized apoptotic tumor cells were absolute requirements. As in clinical ECP, immunogenic cell death of tumor cells was finely titrated by DNA cross-linkage mediated by photoactivated 8-methoxypsoralen (8-MOPA). ECP-induced tumor-loaded DC were effective immunotherapeutic agents only if they were spared exposure to 8-MOPA, indicating that healthy DC are required for ECP. Infusion of responder T cells into naïve tumor-challenged mice established the protective role of stimulated T-cell antitumor immunity. Collectively, these results reveal that selective antitumor effects of ECP are initiated by tumor antigen–loaded, ECP-induced DC, which promote potent collaboration between CD4 and CD8 tumor-specific T cells. These mechanistic insights suggest potential therapeutic applicability of ECP to solid tumors in addition to CTCL.
Significance: These findings identify principal cellular contributors to the anticancer immunotherapeutic impact of ECP and suggest this treatment may be applicable to a broad spectrum of immunogenic malignancies. Cancer Res; 78(14); 4045–58. ©2018 AACR.
Introduction
Extracorporeal photochemotherapy (ECP), an immunotherapy widely operative in university centers worldwide, has long presented a tantalizing mechanistic enigma (1, 2). In common with the physiologic immune system with which it appears to collaborate, ECP has bidirectional clinical impact combined with exquisite specificity, selectively immunizing against the malignant cells of cutaneous T-cell lymphoma (CTCL; refs. 3, 4), while selectively tolerizing to targeted antigens in the transplant setting (5, 6). The immunogenicity and specificity of ECP are highlighted by its dependency on an intact CD8 T-cell compartment in CTCL (7) and by its highly favorable adverse reaction profile with no known increased susceptibility to opportunistic infections in either CTCL or transplant settings.
ECP's ex vivo initiation of monocyte-to-antigen–presenting dendritic cell (DC) maturation suggests that ECP-induced DC may mediate its clinical responses. ECP-produced DC efficiently internalize, process, and cross-present antigens derived from 8-MOPA apoptosis of leukemic, or autoreactive, leukocytes (8). Platelets, following activation in the ECP plate, signal the monocyte-to-DC maturation (9), which then capably cross-present internalized tumor antigens to activate antigen-specific CD8 T cells (10).
Hope that improved understanding of ECP's mechanism might expand its therapeutic reach to a broader spectrum of cancers has generated an international hunt. To accelerate identification of the principal cellular contributors to ECP's anticancer and tolerogenic effects, two nationally sanctioned workshops were conducted and reported: an NIH State-of-the-Science symposium (1) and an American Society for Apheresis consensus conference (11).
Standard ECP treatment, delivered on an outpatient basis, involves passage of the patient's leukapheresis-enriched mononuclear leukocytes through a 1-mm-thick ultraviolet A light (UVA)–transparent plastic plate (1, 2). In the plate, the leukocyte suspension is exposed to UVA-activated 8-methoxypsoralen (8-MOP), which transiently converts the drug to a reactive DNA cross-linking form (8-MOPA), via its bivalent binding to pyrimidine bases on sister DNA strands (12, 13). The processed leukocytes are then returned intravenously to the patient. Because neither the return of leukapheresed blood (14), nor the local activation of 8-MOP in skin infiltrates of CTCL patients by exposure of their integument to ultraviolet irradiation (15), produces comparable immunotherapeutic effects, it is passage of leukocytes through the plastic UVA-exposure plate in conjunction with 8-MOPA that produces cellular alterations critical to clinical response.
Despite abundant published reports addressing ECP's mechanism, two large hurdles have impeded scientific advances. First, lack of a miniaturized device that reproduces ECP's cellular and in vivo effects has precluded experimental animal elucidation of the roles of individual variables. Second, availability of ECP-processed patient leukocytes for in-depth laboratory analysis has been logistically limited by accessibility to treatment centers, timing of the infusions, and ethical need for uncompromised sets of reinfused leukocytes.
We report here the development and experimental use of a miniaturized scalable mouse-to-man ECP device that reproduces both the cellular and in vivo effects of the human ECP device. After first establishing the capacity of this system to reliably produce a tumor-specific antimelanoma effect in mice with established tumors, we then subtracted individual candidate cellular contributors from the inoculum. Our findings reveal that ECP-induced DC, loaded with apoptotic malignant cells, sort the derived tumor antigens and present them to collaborating recipient CD4 and CD8 T cells, to generate the tumor-specific responses. Because parallel, non–cross-reactive responses were similarly created to a syngeneic colorectal cancer, these results suggest potential application of this treatment to management of a broader array of immunogenic cancers.
Materials and Methods
Animals
C57BL/6J mice were purchased from the Jackson Laboratory. Age- and sex-matched mice that were at least 4 weeks of age were used for all experiments. Experiments were performed according to the Yale Institutional Animal Care and Use Committee–approved animal protocols, in agreement with the National Institutes of Animal Healthcare Guidelines. Mice were maintained under specific pathogen-free conditions, food, and water provided ad libitum. The animal facility is accredited by the Association for Assessment of Laboratory Animal Care.
Tumor cell lines
YUMM1.7 melanoma cells were kindly provided by Dr. Bosenberg, Yale University, New Haven, CT (16). MC38 cells (17) were generously provided by Dr. Richard Hynes, MIT, with permission of Dr. Ajit Varki, University of California, San Diego, La Jolla, CA. Cells were authenticated by meticulous record keeping and vial labeling at the providing laboratories and subsequently at our laboratory. All cell lines are routinely tested for Mycoplasma by standard PCR methods at each cell batch freezing. Cells were used for experiments within one passage of thawing a frozen aliquot. All cells were cultured in Dulbecco's Modified Eagle's nutrient mixture F-12 (Ham) medium (DMEM/F12; Gibco) supplemented with 10% heat-inactivated FBS (Atlanta Biologicals), 1% penicillin/streptomycin (Invitrogen), and 1% NEAA (Invitrogen), under standard conditions.
Mouse tumor studies
For tumor induction, 1 × 105 YUMM1.7 or MC38 tumor cells were injected subcutaneously in 100 μL into the right flanks of recipient wild-type C57BL/6J mice. Tumor volume was monitored via biweekly measurement of perpendicular tumor diameters and height using a caliper, and tumor volume calculated as tumor length × width × height/2. In order to combine data from multiple repeat experiments, for each individual experiment, tumor volumes were internally normalized to the average final volume of PBS control tumors in that experiment. The mean relative tumor volumes were then calculated for the cumulative data. Differences in tumor volume were statistically assessed using Sidak multiple comparisons test.
Tumors were harvested in their entirety for histology at the end of some experiments. Although the investigators assessing tumor growth were not blinded, all pathologists and laboratory personnel who performed histologic characterizations of tumor specimens were blinded to group allocation.
8-MOPA treatment of tumor cells
To induce apoptosis, 2.5 × 106 tumor cells were incubated with 100 ng/mL (for YUMM1.7 cells) or 600 ng/mL (for MC38 cells) of 8-MOP (UVADEX; Therakos) in 300 μL of FBS for 20 minutes, and subsequently exposed to 4 J/cm2 (for YUMM1.7 cells) or to 7 J/cm2 (for MC38 cells) of UVA irradiation. Of note, 8-MOP and UVA dosages for each cell line were preestablished by titration, to identify the minimal 8-MOPA dose required to render tumor cells nonviable (tumor cell viability assessed by monitoring cell survival and proliferation over 10-day culture following 8-MOPA exposure).
Isolation of murine peripheral blood mononuclear cells
Peripheral blood was collected from experimental and control mice twice a week for 3 weeks via nonlethal bleeding of the superficial temporal vein. Whole blood was collected into 1:100 5,000 U/mL heparin (McKesson Packaging Services). Platelet-containing peripheral blood mononuclear cells (PBMC) were isolated from peripheral whole blood via Lympholyte M gradient separation (Cedarlane Labs) followed by treatment with ACK buffer (Lonza) to eliminate the residual red blood cells. Autologous platelet-containing plasma was collected and reserved for subsequent steps.
Transimmunization chamber
The miniaturized ECP device suitable for work in animal models, called the Transimmunization (TI) chamber, was designed and created for Dr. Edelson's laboratory by Transimmune AG in collaboration with Fraunhofer Institute for Biomedical Engineering, Saarland, Germany. The sterile polystyrene TI chamber has the external dimensions of 25 × 75 mm, with the flow path of 18 × 66 mm, and the flow passage height of 290 ± 15 μm.
Mouse TI treatment protocol
8-MOPA–treated YUMM1.7 or MC38 tumor cells were combined 1:1 with isolated murine platelet-containing PBMC in FBS. In order to simulate the first five cycles of ECP (which involve cell collection over ∼1 hour), the cell mixture was incubated in the TI chamber for 1 hour at 37°C. This step allows for platelet-activating plasma protein deposition in the chamber, and platelet adherence to the coated chamber surfaces, as confirmed by light microscopy. To simulate ECP plate passage, the mixed cells were subsequently passed through the chamber using a syringe pump, at a rate of 0.09 mL/min. Following plate passage, cells were collected, and the TI chamber washed with 100% FBS at 0.49 mL/min while being physically perturbed by flicking or tapping the plate surface to help detach any adherent cells from the chamber. The collected cells were washed and cultured overnight at standard conditions in RPMI without phenol red (Gibco) supplemented with 15% autologous mouse plasma and 1% penicillin/streptomycin (Invitrogen). The following day, cells were harvested by scraping, washed, resuspended in reserved autologous mouse plasma, and administered intravenously via the retro-orbital plexus.
In experiments where only PBMC or only YUMM1.7 cells were used, the protocol was performed as described above, including 1-hour cell incubation in the TI chamber, plate passage, and overnight incubation prior to cell reinfusion, but omitting either the 8-MOPA–treated YUMM1.7 cells or the PBMC, respectively. In experiments omitting the TI chamber passage step, mixed PBMC and 8-MOPA–treated YUMM1.7 cells were immediately put into overnight coculture (as described) following cell mixture. In P-selectin–blocking experiments, PBMC/YUMM1.7 cell suspensions were incubated with 100 μg of purified anti–CD62P-blocking antibody (clone RB40.34, BD Biosciences) for 5 minutes prior to plate passage, with no additional wash steps before cell passage through the TI chamber.
Ex vivo depletions from PBMC
CD11b monocyte depletion.
Column-free magnetic separation was performed on the PBMC using the EasySep mouse CD11b-positive selection kit (STEMCELL Technologies) prior to TI chamber incubation with the YUMM1.7 cells.
CD41 platelet depletion.
Column magnetic separation was performed on the PBMC using the CD41 Biotin and anti-Biotin MicroBeads (Miltenyl Biotec) prior to TI chamber incubation with the YUMM1.7 cells.
8-MOPA treatment of PBMC
In experiments where PBMC were exposed to 8-MOPA, 2.5 × 106 freshly isolated PBMC were incubated with 100 ng/mL of 8-MOP in 300 μL of FBS for 20 minutes and subsequently exposed to 4 J/cm2 of UVA irradiation. The 8-MOPA–treated PBMC were then either used directly in the standard TI protocol described above, as the sole PBMC source, or added to the standard TI protocol together with 8-MOPA–spared PBMC as a 1:1 mix.
PKH phagocytic assay
In order to determine the phagocytic potential of TI-treated monocytes, YUMM1.7 tumor cells (mouse) or YuCOT primary melanoma cells (human) were labeled with the membrane protein dye PKH-red (Sigma) per the manufacturer's instructions. 8-MOPA–treated tumor cells were then combined with respectively mouse or human PBMC and either underwent plate passage per TI protocol, or TI protocol with omission of plate passage, with platelet depletion, or with P-selectin blocking. PBMC and labeled tumor cells were incubated overnight as outlined above. The next day, cells were harvested, washed, resuspended in FACS buffer (PBS supplemented with 2% FBS), treated with Fcγ blocker for 5 minutes, and stained with fluorescently conjugated antibodies. Anti-CD11b (eBiosciences, clone M1/70), anti-CD19 (eBiosciences, clone 6D5), and anti-MHCII (eBiosciences, clone M5/114.15.2) antibodies were used to identify phagocytic cells in mouse PBMC, and CD11c (BioLegend, clone 3.9) in human PBMC, within which cells' PKH fluorescence was monitored via the PE channel. Flow cytometry was done with a Stratedigm flow cytometer, and data were analyzed with FlowJo software (FlowJo LLC).
Mouse Luminex cytokine analysis
Following the overnight incubation step of the TI protocol, 100 μL of culture supernatant was harvested from individual cell cultures, centrifuged to remove any contaminating cells or debris, and stored at −20°C. Collected supernatants were subsequently screened for the presence of 23 different cytokines and chemokines (IL1a, IL1b, IL2, IL3, IL4, IL5, IL6, IL9, IL-10, IL12p40, IL12-p70, IL13, IL17, Eotaxin, G-CSF, GM-CSF, IFNγ, KC, MCP-1, MIP-1a, MIP-1b, RANTES, and TNFα) by Yale University Immune Monitoring Core Facility for Luminex testing.
Mouse in vivo immune cell subset depletions by antibody injection
Mice were injected intraperitoneally on day 10, just prior to first TI treatment, with 500 μg/mouse of anti-NK1.1 (Biolegend, clone DK136), 100 μg/mouse anti-CD8 (Biolegend, clone 53-6.7), or with 100 μg/mouse anti-CD4 (Biolegend, clone GK1.5) antibody per the manufacturer's protocol. Injections were repeated biweekly for a-CD4 and a-CD8 treatments and weekly for a-NK1.1 treatments, for the duration of the experiment, just prior to each treatment. Depletion was confirmed by longitudinal FACS analysis of relevant cell subsets in peripheral blood of immune-depleted animals.
Spleen adoptive transfer assays
TI-treated and control tumor-bearing mice were sacrificed within 1 week of their 6th and final treatment, and splenocytes were collected. Spleens were also collected from untreated, tumor-free, age- and gender-matched naïve mice. Splenocytes were prepared by homogenizing the tissue by passage through a nylon mesh (70 μm) and density centrifugation over Lympholyte M. For whole splenocyte transfers, splenocytes were resuspended in PBS and injected via the retro-orbital plexus in naïve 5-week-old male C57BL/6J mice. One donor spleen equivalent per recipient mouse was injected. For CD3-enriched cell transfers, CD3+ cells were isolated from whole splenocytes by column magnetic separation (Miltenyi Biotec), and then injected as described above. Mice receiving either whole splenocyte or enriched splenic CD3 T-cell transfers were challenged with a tumor inoculum of 1 × 105 YUMM1.7 cells, injected subcutaneously into the right flank on the day of splenocyte transfer. Tumor volumes were measured, data normalized, and statistical significance calculated as described above for tumor growth experiments.
Splenic CD4 T-cell IFNγ assay and MHCII blockade
In order to determine if there was evidence of CD4 activation associated with TI, recipients of spleen transfer from TI mice. In addition, splenocytes were collected from tumor-bearing control mice that have not received any splenocyte transfer (“untreated control”). All mice were sacrificed after final tumor measurements, approximately 30 days following tumor inoculation. Spleens were harvested as noted above. CD11b+ cells and CD4+ T cells were isolated from splenocytes via negative selection with STEMCELL isolation kits. Purified cells were plated overnight either alone or together for 3 or 6 days in T-cell medium at 1 × 105 cells/well in a 96-well plate. For MHCII blocking, 1 μg/well of anti-MHCII I-A/I-E antibody (Biolegend) was added to the appropriate wells and renewed every other day for the duration of the experiment. Supernatants from the cultures were collected and sent for IFNγ analysis by Luminex.
Human PBMC isolation and TI treatment protocol
All studies were performed with blood donated by healthy volunteers. We obtained written informed consent from all volunteers, the studies were conducted in accordance with recognized ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, U.S. Common Rule), and the studies were approved by Yale Human Investigational Review Board under protocol number 0301023636.
Peripheral blood was collected into 1:100 5,000 U/mL heparin (McKesson Packaging Services), and platelet-containing PBMC isolated by density gradient centrifugation over Isolymph (CTL Scientific Supply Corp.) following the manufacturer's protocol. Autologous plasma (also containing platelets) was collected and reserved. Washed PBMC and platelets were resuspended in autologous plasma and incubated for 1 hour either in the TI chamber or in the clinical ECP plate. The cells were then either passed through the TI chamber under conditions described above for mouse TI protocol or circulated for 1 hour though the clinical ECP plate at the flow rate of 24 mL/min, followed by a 100 mL/min wash with human AB serum (Lonza BioWhittaker) with physical perturbation by flicking or tapping the plate surface, to help detach any adherent cells. PBMC passed through either the TI chamber or the ECP plate were collected, washed, and cultured overnight under standard conditions in RPMI without phenol-red (Gibco) supplemented with 15% Human AB serum (Lonza BioWhittaker), 1% penicillin/streptomycin (Invitrogen), and 1% l-glutamine (Invitrogen). The following day, cells were harvested (including harvest of any attached cells by scraping) for analysis.
Human PBMC FACS analysis
Cells harvested after overnight incubation (described above) were stained following standard protocols. Intracellular staining was performed using the IntraPrep Permeabilization Reagent (Beckman Coulter #A07803). The experiments used antibodies specific for human CD83 (HB15E), CD80 (2D10), CD14 (M5E2), CD11c (3.9), CD112 (TX31), ICAM1 (ha58), CXCL5 (J111B7), and Tnfrsf10B (DJR2-4(7-8)) obtained from BioLegend; CD86 (2331) and TNFSF9 (C65-485) purchased from BD Biosciences; HLA-DR (B8.12.2) from Beckman-Coulter; PLAUR (vim5), SIRPa (15-414), CD105 (sn6), HLA-DR (LN3), and MCP1 (5D3-F7) obtained from eBiosciences; ITGB8 (416922) from Invitrogen; ABCA1 (hj1), ITGB5 (238307), and Tnfsf14 (MM0456-8C34) from Novus Biologicals; and MSR1 (351615), CXCL16 (256213), ORL1 (331212), and TNFRSF1A (16803) from R&D Systems. Color-matched isotype control antibodies were obtained from the same vendors.
Flow cytometry was done with a Stratedigm flow cytometer with electronic gates set on live cells by a combination of forward and side light scatter and 7-AAD (BioLegend) exclusion. A minimum of 3 × 104 events were collected per sample, and data were analyzed with FlowJo software (FlowJo LLC). Change in mean fluorescence intensity (ΔMFI) was calculated by subtracting the MFI of corresponding IgG control sample from MFI of the antibody-stained sample. Percent (%) change in positively expressing cells was calculated in similar fashion. Statistical significance in comparisons was calculated using the paired t test analysis.
Results
Experimental ECP protocol controls murine syngeneic tumor growth
ECP's bidirectionality (induction of both anticancer immunity and tolerance to transplants) has complicated its mechanistic dissection. To focus on its anticancer impact, we established a murine ECP model of immunization against solid syngeneic tumors. To do so, we developed a scalable mouse-to-man micro-ECP device, the TI chamber (Fig. 1A), which replicates key features of ECP, and designed relevant treatment protocols. Since our earlier studies had revealed that ECP's tolerogenic effect is mediated by 8-MOPA–injured antigen-presenting cells (18), we polarized experimental ECP toward immunization mode by sparing monocyte/DC exposure to 8-MOPA. Furthermore, because ECP's immunotherapeutic specificity is clinically enhanced (19) by maximizing contact between 8-MOPA–injured malignant cells and ECP-induced DC, we incorporated an overnight incubation step between these two cell types before introduction of the tumor-loaded DC. For clarification, we have entitled ECP's immunization mode as “Transimmunization” and its tolerogenic mode as “Transtolerization.” This study focuses on the former.
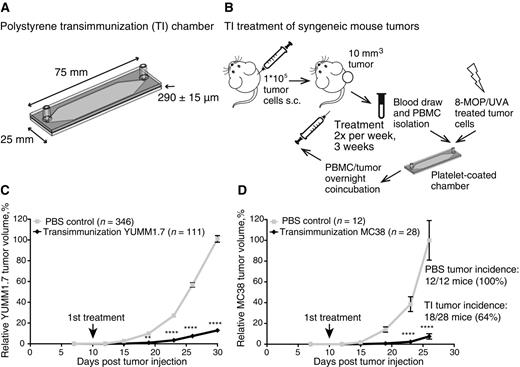
TI treatment significantly reduces growth of mouse syngeneic tumors. A, Diagram and specifications of TI treatment chamber. B, Schematic description of TI treatment experimental workflow. Briefly, animals are inoculated subcutaneously with syngeneic tumor cells; animals with palpable tumors are treated twice weekly by blood draw, isolation of PBMC from blood, PBMC flow passage through the autologous platelet-coated TI chamber in the presence of 8-MOP/UVA-treated tumor cells, PBMC and tumor cell coincubation overnight, and reinjection of the cells intravenously into the same tumor-bearing animals. Tumor volume is measured throughout the experiment. C, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six TI treatments (TI), or bled on the same schedule but receiving six sterile PBS reinfusions (PBS). Arrow, first treatment day. Data are cumulative over nine independent experiments. D, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 MC38 tumor cells; treatments as in C. Arrow, first treatment day. Data are cumulative over two independent experiments. Tumor incidence defined by tumor volume >5 mm3 (tumor) or <5 mm3 (no/regressed tumor) at the final measurement. C and D, For each individual experiment, tumor volumes were normalized to the average final volume of PBS control tumors for that experiment. The mean relative tumor volumes were then calculated for the cumulative data. Error bars, SEM. P values calculated for each time point using Sidak multiple comparisons test; **, P = 0.0013 and ****, P < 0.0001.
TI treatment significantly reduces growth of mouse syngeneic tumors. A, Diagram and specifications of TI treatment chamber. B, Schematic description of TI treatment experimental workflow. Briefly, animals are inoculated subcutaneously with syngeneic tumor cells; animals with palpable tumors are treated twice weekly by blood draw, isolation of PBMC from blood, PBMC flow passage through the autologous platelet-coated TI chamber in the presence of 8-MOP/UVA-treated tumor cells, PBMC and tumor cell coincubation overnight, and reinjection of the cells intravenously into the same tumor-bearing animals. Tumor volume is measured throughout the experiment. C, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six TI treatments (TI), or bled on the same schedule but receiving six sterile PBS reinfusions (PBS). Arrow, first treatment day. Data are cumulative over nine independent experiments. D, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 MC38 tumor cells; treatments as in C. Arrow, first treatment day. Data are cumulative over two independent experiments. Tumor incidence defined by tumor volume >5 mm3 (tumor) or <5 mm3 (no/regressed tumor) at the final measurement. C and D, For each individual experiment, tumor volumes were normalized to the average final volume of PBS control tumors for that experiment. The mean relative tumor volumes were then calculated for the cumulative data. Error bars, SEM. P values calculated for each time point using Sidak multiple comparisons test; **, P = 0.0013 and ****, P < 0.0001.
The proof-of-principle murine TI protocol (Fig. 1B) consists of extracorporeal passage of PBMC from tumor-bearing mice through the TI chamber, in the presence of apoptotic malignant cells generated through exposure to photoactivated 8-methoxypsoralen (8-MOPA). The TI chamber–activated immune cells are coincubated overnight with the 8-MOPA–damaged dying tumor cells, facilitating tumor cell phagocytosis and transfer of “personalized” tumor antigens to the DC. On the following day, the coincubated cell mixture is reinfused intravenously into the tumor-bearing animal. To establish the protocol's efficacy against established cancer, immunotherapy was initiated after subcutaneously injected syngeneic tumors become palpable. The protocol was repeated twice weekly for 3 weeks, for a total of six treatments. Control animals underwent blood collection on the same schedule, to normalize for any effects of lymphodepletion on tumor growth, but received PBS reinfusions. Tumor growth was monitored throughout the experiment. The treatment appeared well tolerated in all animals treated (>500).
To test the efficacy of TI, we utilized the YUMM1.7 murine melanoma (16) and the MC38 colon carcinoma (17) syngeneic systems. The systems were selected to represent tumors of different tissue origins (skin vs. gut) and of different mutational burden status. YUMM1.7 cells are derived from a relatively mutation-poor genetically engineered tumor model (310 exonic coding mutations, not filtered for expression; M. Bosenberg, personal communication), whereas MC38 cells are derived from a mutation-rich chemically induced tumor model (4285 exonic coding mutations, ref. 20). TI-treated mice experienced a highly significant delay in tumor growth as compared with untreated mice in both the YUMM1.7 and the MC38 tumor models, demonstrating remarkable and consistent efficacy of the protocol against solid tumors (Fig. 1C and D; Supplementary Fig. S1A and S1B). Intriguingly, although the growth of mutation-poor YUMM1.7 tumors was controlled but not prevented by TI treatment (Fig. 1C; Supplementary Figs. S1A–S1B, S2, and S3A–S3B), in the more mutation-rich MC38 model, the tumor incidence was also affected, with complete regression of up to 30% of inoculated tumors (Fig. 1D). These data support the antitumor effectiveness of the ECP-derived TI protocol in multiple tumor types and extend the potential applications of modified ECP from CTCL to solid tumors.
TI selectivity is dependent on apoptotic tumor antigen source
High selectivity for the disease-causing cells has been a hallmark of ECP treatment in its 20+ years of clinical use (2). In treatment of CTCL, the antitumor immunity stimulated by ECP is specific to the malignant cell clone, with no concomitant autoimmune or nonspecific side effects (2, 4). When using the TI protocol in mice, we similarly did not observe any signs of general autoimmunity or of inappropriate epitope spreading to healthy tissue of similar origin as the tumor (such as hair graying or vetiligo in the YUMM1.7 system, or colitis in the MC38 system).
To test more rigorously whether this key aspect of ECP is conserved in TI treatment, and to confirm the antigen specificity of the observed antitumor responses, we cross-treated YUMM1.7 tumor–bearing mice with MC38-loaded autologous leukocytes, and, conversely, treated MC38 tumor–bearing mice with YUMM1.7-loaded autologous leukocytes. As predicted by the antigen specificity associated with clinical ECP, we did not observe any cross-protection against YUMM1.7 tumors with MC38 cells as the antigen source, or against MC38 tumors with YUMM1.7 cells as antigen (Fig. 2A). These data indicate that, similar to ECP, TI treatment in mouse solid tumor models is antigen-specific and linked to antigen transfer from malignant cells to stimulatory DC. This conclusion was confirmed in additional experiments, which introduced the antigen source (8-MOPA–treated tumor cells) or TI-treated leukocytes separately, with neither component showing therapeutic efficacy in isolation (Fig. 2B; Supplementary Fig. S4).
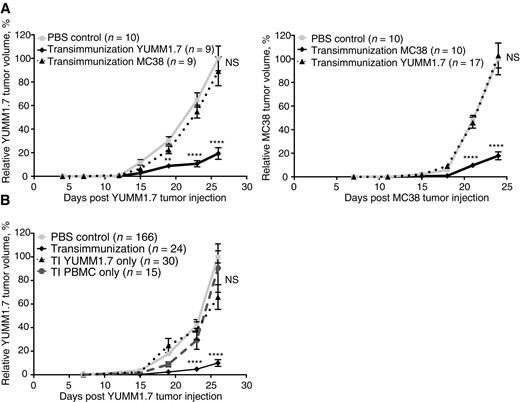
TI is specific and requires both TI-treated immune cells and 8-MOP/UVA-exposed tumor cells. A, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with either 1 × 105 YUMM1.7 or MC38 tumor cells and receiving six TI treatments using either the same (solid black lines) or different (dotted lines) tumor cells than the primary tumor. “PBS control” mice were bled on the same schedule but received six sterile PBS reinfusions. B, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six full treatments (TI), six reinfusions of TI-treated PBMC alone without tumor cells (TI PBMC only), or six reinfusions of 8-MOP/UVA-exposed tumor cells alone without PBMC (TI YUMM1.7 only). “PBS control” mice were bled on the same schedule but received six sterile PBS reinfusions. Data are cumulative over three independent experiments. A and B, For each experiment, individual tumor volumes were normalized to the average final volume of PBS control tumors for that experiment, and mean relative tumor volumes were then calculated. Bars, SEM. P values calculated for each time point using Sidak multiple comparisons test; **, P = 0.0011; ****, P < 0.0001; NS, differences not significant.
TI is specific and requires both TI-treated immune cells and 8-MOP/UVA-exposed tumor cells. A, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with either 1 × 105 YUMM1.7 or MC38 tumor cells and receiving six TI treatments using either the same (solid black lines) or different (dotted lines) tumor cells than the primary tumor. “PBS control” mice were bled on the same schedule but received six sterile PBS reinfusions. B, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six full treatments (TI), six reinfusions of TI-treated PBMC alone without tumor cells (TI PBMC only), or six reinfusions of 8-MOP/UVA-exposed tumor cells alone without PBMC (TI YUMM1.7 only). “PBS control” mice were bled on the same schedule but received six sterile PBS reinfusions. Data are cumulative over three independent experiments. A and B, For each experiment, individual tumor volumes were normalized to the average final volume of PBS control tumors for that experiment, and mean relative tumor volumes were then calculated. Bars, SEM. P values calculated for each time point using Sidak multiple comparisons test; **, P = 0.0011; ****, P < 0.0001; NS, differences not significant.
Together, the observed selectivity of TI treatment for tumor cell type and the requirement for both TI-activated immune cells and 8-MOPA-treated tumor cells are consistent with an immune response initiated by antigen-presenting cells.
Monocytes, platelets, and TI chamber are necessary for TI antitumor efficacy
Previous research into the mechanisms underlying the therapeutic efficacy of ECP has indicated a critical role for blood monocyte–derived DC, which are physiologically activated by interactions with platelets under flow conditions in the clinical ECP chamber (9). The micro-sized TI chamber experimental system for the first time has allowed accurate dissection of the complex and overlapping factors leading to DC creation and activation in ECP.
To this end, experiments were designed to individually test the contribution of the system's components. The depletion of the monocyte fraction from PBMC prior to passage through the TI chamber completely removed the therapeutic effect of TI treatment on YUMM1.7 tumors (Fig. 3A; Supplementary Fig. S5A), confirming that monocytes are critical. We then sought to define the phenotypic and physiologic consequences of monocyte ex vivo interaction with the TI chamber. Although the treatment did not significantly affect the ability of monocytes to take up apoptotic 8-MOPA–treated YUMM1.7 cells (Fig. 3B), it induced a distinct inflammatory cytokine secretion profile, characterized most prominently by increased IL6 and CCL2 (MCP-1; Fig. 3C; Supplementary Fig. S5B). This cytokine profile was dependent on the presence of monocytes, as monocyte depletion largely abrogated it (Fig. 3C). It was also abrogated by the omission of the TI chamber passage step from the protocol (Fig. 3C) and by depletion of platelets prior to plate passage (Supplementary Fig. S5C). These data suggest that TI treatment activates monocytes in a unique manner that requires the TI chamber as well as the presence of platelets in the chamber. In previous work, platelet P-selectin has been demonstrated to contribute to monocyte activation by ECP (9). In addition, CCL2 has been specifically linked to P-selectin–dependent monocyte activation (21, 22). We confirmed that in our system, blocking P-selectin also abrogates the inflammatory cytokine profile (Fig. 3D), supporting the hypothesis that activated platelets contribute to monocyte activation through this pathway.
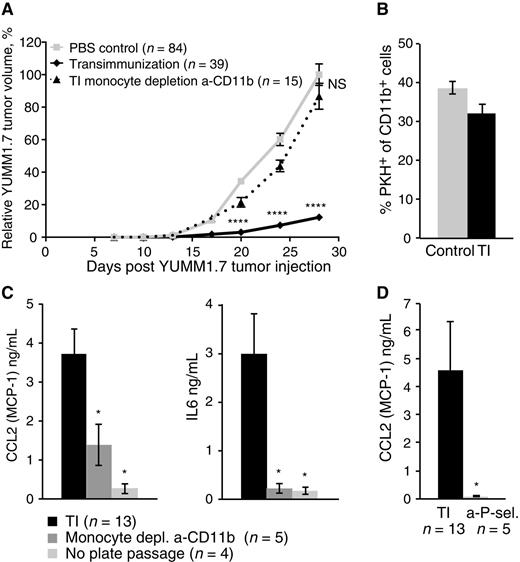
Monocytes are necessary for TI and contribute to a distinct in vitro inflammatory cytokine profile that is also dependent on plate passage and platelet activation. A, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six TI treatments (solid black lines) or six TI treatments where monocytes were depleted from PBMC prior to plate passage step using anti-CD11b depletion kit (dotted lines). “PBS control” mice were bled on the same schedule but received six sterile PBS reinfusions. Data are cumulative over three independent experiments. For each experiment, individual tumor volumes were normalized to the average final volume of PBS control tumors for that experiment, and mean relative tumor volumes were then calculated. Bars, SEM; P values calculated for each time point using Sidak multiple comparisons test; ****, P < 0.0001; NS, differences not significant. B, FACS analysis of phagocytic potential of CD11b+ cells within PBMC that have either been freshly isolated (control) or TI-treated (TI) prior to overnight coincubation with PKH-labeled, 8-MOP/UVA-exposed YUMM1.7 tumor cells. Bars represent average % PKH+ cells of CD11b+ cells, measured in three independent experiments. Error bars, SEM. C, Luminex analysis of CCL2 (MCP-1) and IL6 concentrations in the overnight culture supernatants collected in the course of standard TI experiments, or TI experiments with the indicated changes (a-CD11b kit monocyte depletion prior to plate passage, or omission of plate passage step). D, Luminex analysis of CCL2 (MCP-1) concentration in the overnight culture supernatants collected in the course of standard TI experiments, or TI experiment adding an a-P-selectin antibody–blocking step prior to PBMC plate passage. C and D, Bars represent mean cytokine concentrations, measured in the indicated number of independent experiments. Error bars, SEM; P values calculated using the Student t test; *, P < 0.05.
Monocytes are necessary for TI and contribute to a distinct in vitro inflammatory cytokine profile that is also dependent on plate passage and platelet activation. A, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six TI treatments (solid black lines) or six TI treatments where monocytes were depleted from PBMC prior to plate passage step using anti-CD11b depletion kit (dotted lines). “PBS control” mice were bled on the same schedule but received six sterile PBS reinfusions. Data are cumulative over three independent experiments. For each experiment, individual tumor volumes were normalized to the average final volume of PBS control tumors for that experiment, and mean relative tumor volumes were then calculated. Bars, SEM; P values calculated for each time point using Sidak multiple comparisons test; ****, P < 0.0001; NS, differences not significant. B, FACS analysis of phagocytic potential of CD11b+ cells within PBMC that have either been freshly isolated (control) or TI-treated (TI) prior to overnight coincubation with PKH-labeled, 8-MOP/UVA-exposed YUMM1.7 tumor cells. Bars represent average % PKH+ cells of CD11b+ cells, measured in three independent experiments. Error bars, SEM. C, Luminex analysis of CCL2 (MCP-1) and IL6 concentrations in the overnight culture supernatants collected in the course of standard TI experiments, or TI experiments with the indicated changes (a-CD11b kit monocyte depletion prior to plate passage, or omission of plate passage step). D, Luminex analysis of CCL2 (MCP-1) concentration in the overnight culture supernatants collected in the course of standard TI experiments, or TI experiment adding an a-P-selectin antibody–blocking step prior to PBMC plate passage. C and D, Bars represent mean cytokine concentrations, measured in the indicated number of independent experiments. Error bars, SEM; P values calculated using the Student t test; *, P < 0.05.
Having established the importance of TI chamber and activated platelets in ex vivo monocyte activation, we next investigated the effect of these components on the in vivo therapeutic efficacy of the TI protocol. We find that, consistent with the in vitro effects, either omitting the TI chamber passage step, or depleting platelets prior to plate passage, completely abrogates antitumor efficacy of TI against YUMM1.7 (Fig. 4A and B; Supplementary Fig. S6A and S6B). Together with monocyte depletion results (Fig. 3A), these findings confirm that monocyte interactions with platelets during cell passage through the chamber are key positive regulators of antitumor immunity in this system.
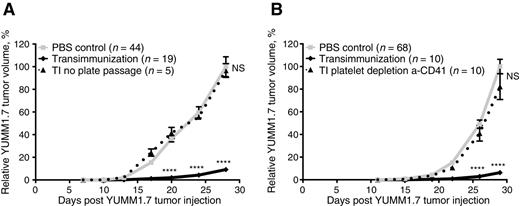
TI requires plate passage in the presence of platelets. A, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six TI treatments (solid black lines) or six TI treatments with plate passage step omitted (dotted lines). B, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six TI treatments (solid black lines) or six TI treatments where platelets were depleted from the PBMC prior to plate passage step using anti-CD41 depletion kit (dotted lines). A and B, “PBS control” mice in all experiments were bled on the same schedule as the experimental animals, but received six sterile PBS reinfusions. Data are representative of at least two independent experiments. For each experiment, individual tumor volumes were normalized to the average final volume of PBS control tumors for that experiment, and mean relative tumor volumes were then calculated. Bars, SEM. P values calculated for each time point using Sidak multiple comparisons test; ****, P < 0.0001 and NS, differences not significant.
TI requires plate passage in the presence of platelets. A, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six TI treatments (solid black lines) or six TI treatments with plate passage step omitted (dotted lines). B, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six TI treatments (solid black lines) or six TI treatments where platelets were depleted from the PBMC prior to plate passage step using anti-CD41 depletion kit (dotted lines). A and B, “PBS control” mice in all experiments were bled on the same schedule as the experimental animals, but received six sterile PBS reinfusions. Data are representative of at least two independent experiments. For each experiment, individual tumor volumes were normalized to the average final volume of PBS control tumors for that experiment, and mean relative tumor volumes were then calculated. Bars, SEM. P values calculated for each time point using Sidak multiple comparisons test; ****, P < 0.0001 and NS, differences not significant.
Conversely, 8-MOPA injury of monocytes has been suggested as a negative regulator of ECP immunity. We confirm that uniform 8-MOPA exposure of the leukocyte fraction completely abrogates the TI protocol efficacy against YUMM1.7 tumors (Fig. 5A; Supplementary Fig. S7A). Preliminary data also suggest that the presence of an equal fraction of 8-MOPA–exposed leukocytes among the 8-MOPA–protected, immunizing TI-leukocytes, significantly reduces the antitumor treatment efficacy (Fig. 5B; Supplementary Fig. S7B). Together, these data support previous hypotheses of ECP mechanism and provide important insight into the key positive and negative regulators of ECP therapy, with implications for intelligent ECP redesign in cancer immunotherapy and tolerogenic applications.

TI is inhibited by immune cell exposure to 8-MOP/UVA, or addition of 8-MOP/UVA-treated PBMC to tumor-protective protocol. A, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six TI treatments (solid black line) or six TI treatments where PBMC were uniformly exposed to 8-MOP/UVA irradiation immediately after plate passage (dotted line). B, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six TI treatments (solid black line) or six treatments where TI cells after plate passage were mixed 1:1 with an equal number of PBMC uniformly exposed to 8-MOP/UVA irradiation (dotted line). A and B, “PBS control” mice in all experiments were bled on the same schedule as the experimental animals, but received six sterile PBS reinfusions. Data are representative of at least two independent experiments. For each experiment, individual tumor volumes were normalized to the average final volume of PBS control tumors for that experiment, and mean relative tumor volumes were then calculated. Bars, SEM. P values calculated for each time point using Sidak multiple comparisons test; **, P < 0.01; ****, P < 0.0001; NS, differences not significant.
TI is inhibited by immune cell exposure to 8-MOP/UVA, or addition of 8-MOP/UVA-treated PBMC to tumor-protective protocol. A, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six TI treatments (solid black line) or six TI treatments where PBMC were uniformly exposed to 8-MOP/UVA irradiation immediately after plate passage (dotted line). B, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving either six TI treatments (solid black line) or six treatments where TI cells after plate passage were mixed 1:1 with an equal number of PBMC uniformly exposed to 8-MOP/UVA irradiation (dotted line). A and B, “PBS control” mice in all experiments were bled on the same schedule as the experimental animals, but received six sterile PBS reinfusions. Data are representative of at least two independent experiments. For each experiment, individual tumor volumes were normalized to the average final volume of PBS control tumors for that experiment, and mean relative tumor volumes were then calculated. Bars, SEM. P values calculated for each time point using Sidak multiple comparisons test; **, P < 0.01; ****, P < 0.0001; NS, differences not significant.
TI activates host innate and adaptive antitumor immunity
Activated DC present tumor antigens in an immune-stimulating context to evoke adaptive and innate immune response against the tumor. In the context of TI, we hypothesize that monocytes stimulated ex vivo by interaction with activated platelets in the TI chamber to become physiologically activated DC, and loaded with apoptotic 8-MOPA–treated tumor cells in overnight coincubation, also proceed to activate host cytotoxic antitumor immunity in vivo.
We therefore investigated whether depletion from the tumor-bearing mice of T-cell subsets, or of natural killer cells, would influence TI treatment outcome of YUMM1.7 tumors. Following systemic depletion of cellular subsets in vivo by injection of depleting monoclonal antibodies, we report that removal of CD8 T cells from the host immediately prior to the first treatment significantly abrogates TI treatment efficacy (Fig. 6A; Supplementary Fig. S8A), consistent with the importance of cytotoxic T cells in antitumor immunity. Interestingly, depletion of CD4 T cells equally diminished the antitumor effect (Fig. 6A; Supplementary Fig. S8A), suggesting that CD4 T cells are also necessary for the observed antitumor immunity, whether directly or indirectly through CD8 T-cell help. In addition, depletion of NK1.1+ cells largely nullified the antitumor response, indicating that cytotoxic innate immunity is activated by TI and plays an important role in this system (Fig. 6B; Supplementary Fig. S8B).

TI requires host innate and adaptive immunity, and creates transferable T-cell antitumor response. A and B, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving six TI treatments (solid black line) alone, or together with systemic depletion of CD8+ or CD4+ (A), or NK1.1+ cells (B), by antibody injection (100 μL of a-CD8 or a-CD4 depleting Ab injected i.p. 2×/week, or 500 μL of a-NK1.1 Ab injected i.p. 1×/week, for the duration of the experiment, beginning on indicated day). “PBS control” mice were bled on the same schedule as the experimental animals, but received six sterile PBS reinfusions. C and D, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and on the same day infused with one spleen equivalent of whole splenocytes (C) or CD3-purified splenic T cells (D) from either tumor-bearing TI-treated mice (solid black lines), tumor-bearing PBS control mice (dotted black lines), or naïve untreated, tumor-free mice (dashed gray lines). Untreated control mice (solid gray lines) received tumor inoculation alone, with no further treatment. A–D, Data in each panel are representative of at least two independent experiments. For each experiment, individual tumor volumes were normalized to the average final volume of PBS control tumors for that experiment, and mean relative tumor volumes were then calculated. Bars, SEM. P values calculated for each time point using Sidak multiple comparisons test; **, P < 0.01; ****, P < 0.0001; NS, differences not significant.
TI requires host innate and adaptive immunity, and creates transferable T-cell antitumor response. A and B, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and receiving six TI treatments (solid black line) alone, or together with systemic depletion of CD8+ or CD4+ (A), or NK1.1+ cells (B), by antibody injection (100 μL of a-CD8 or a-CD4 depleting Ab injected i.p. 2×/week, or 500 μL of a-NK1.1 Ab injected i.p. 1×/week, for the duration of the experiment, beginning on indicated day). “PBS control” mice were bled on the same schedule as the experimental animals, but received six sterile PBS reinfusions. C and D, Mean relative tumor volume over time plotted for C57BL/6 mice inoculated with 1 × 105 YUMM1.7 tumor cells and on the same day infused with one spleen equivalent of whole splenocytes (C) or CD3-purified splenic T cells (D) from either tumor-bearing TI-treated mice (solid black lines), tumor-bearing PBS control mice (dotted black lines), or naïve untreated, tumor-free mice (dashed gray lines). Untreated control mice (solid gray lines) received tumor inoculation alone, with no further treatment. A–D, Data in each panel are representative of at least two independent experiments. For each experiment, individual tumor volumes were normalized to the average final volume of PBS control tumors for that experiment, and mean relative tumor volumes were then calculated. Bars, SEM. P values calculated for each time point using Sidak multiple comparisons test; **, P < 0.01; ****, P < 0.0001; NS, differences not significant.
To confirm that TI treatment leads to bona fide adaptive antitumor immunity, we isolated whole splenocytes (Fig. 6C; Supplementary Fig. S8C) or enriched splenic T cells (Fig. 6D; Supplementary Fig. S8D) either from TI-treated tumor-bearing animals, from tumor-bearing PBS controls, or from tumor-naïve animals. These cells were transferred into naïve mice, which were challenged with YUMM1.7 tumor cells subcutaneously. In this setting, the transfer of either whole unfractionated splenocytes, or of enriched T-cell fractions, from TI-treated mice, uniquely suppressed YUMM1.7 tumor growth.
To further dissect the mechanism of tumor protection by transferred splenocytes, cocultures of CD4+ T cells and CD11b+ monocytes isolated from splenocyte transfer-recipient mice at day 30 after transfer were established. IFNγ was increased in supernatants collected from cocultures of monocytes and CD4+ cells from mice that have received TI splenocyte transfer, but not from similar cocultures of tumor-bearing control mouse cells (Supplementary Fig. S9). The addition of an anti–MHCII-blocking antibody significantly inhibited this TI-specific IFNγ production. This evidence further supports TI induction of antigen-specific T cells by monocyte-derived DC.
Together, these findings suggest that TI is capable of activating a complex interplay of innate and adaptive antitumor immune mechanisms. They also confirm that the TI protocol indeed activates an adaptive, T-cell–mediated antitumor response, which leads to transferrable protection equivalent to active treatment.
TI protocol activates human monocytes into physiologic DC
To investigate whether phenotypic changes observed in the cancer-immunogenic TI-treated monocyte-derived DC would also be observed in human PBMC subjected to similar conditions, we extended the TI procedure to use freshly isolated human PBMC. Normal donor PBMC were either treated with passage through the autologous platelet-coated TI chamber, or circulated for 1 hour through a platelet-coated clinical ECP plate under flow conditions typical of therapeutic ECP. As in murine experiments, PBMC populations were not exposed to 8-MOPA during plate passage. In addition, in an effort to dissect plate-only effects of ECP, PBMC were not exposed to any apoptotic tumor cells during the overnight incubation.
From previous findings of phenotypic markers associated with DC activation by ECP (8), as well as established makers of human DC differentiation, we designed a flow cytometry analysis panel. The panel was applied to samples containing either freshly isolated PBMC, or PBMC following treatment with either the TI chamber or the clinical ECP plate analyzed after overnight incubation. As shown in Fig. 7 and in Supplementary Table S1, exposure of human PBMC to either the TI chamber, or to the clinical ECP plate, robustly and comparably induces markers consistent with DC activation and maturation, such as HLA-DR, CD80, CD83, and CD86 (Fig. 7A), and markers associated with ECP activation of monocytes, such as ICAM-1 and PLAUR (Fig. 7B). Importantly, we also observed inflammatory chemokine expression similar to that characterized in mouse experiments, including upregulation of CCL2 (Fig. 7C). We were also able to demonstrate that, similar to the murine system, this activation of human DC by the TI protocol required cell passage through the activation chamber and the presence of platelets. Removal of either of these significantly reduced activation (Fig. 7D). Also similar to the murine system (Fig. 3B), TI treatment did not significantly affect the ability of human monocytes to take up apoptotic 8-MOPA–treated YuCOT primary melanoma cells (Fig. 7E).
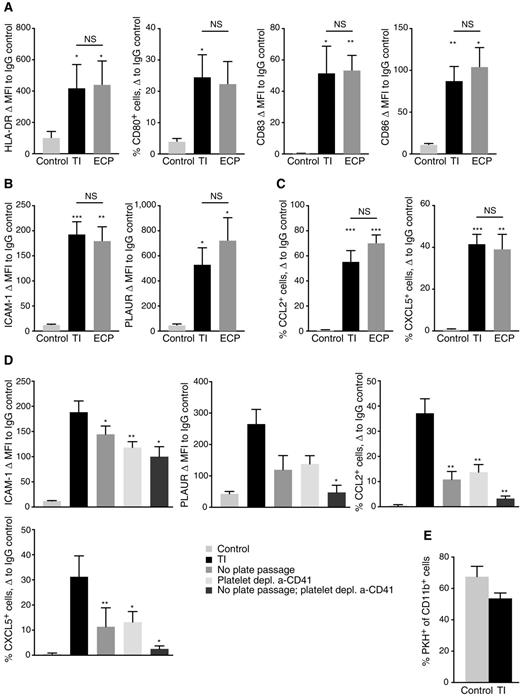
TI protocol with TI chamber or the clinical ECP plate rapidly induces DC maturation and unique activation profile in human PBMC, dependent on plate passage and platelets. A and B, FACS analysis of MFI change of the indicated markers from corresponding IgG controls in live CD11c+ cells among either freshly isolated PBMC (control), PBMC treated with the TI protocol and analyzed following overnight incubation (TI), or PBMC treated with the TI protocol adapted to the clinical ECP plate and analyzed following overnight incubation (ECP). For CD80, where only a subset of cells express the marker, difference in percent CD80+ cells of live CD11c+ PBMC is instead represented. C, FACS analysis of percent change of the indicated markers from corresponding IgG controls in fixed and permeabilized CD11c+ cells among either freshly isolated PBMC (control), PBMC treated with the TI protocol and analyzed following overnight incubation (TI), or PBMC treated with the TI protocol adapted to the clinical ECP plate and analyzed following overnight incubation (ECP). D, FACS analysis of TI-specific marker expression in live CD11c+ cells among either freshly isolated PBMC (control), PBMC treated with the TI protocol and analyzed following overnight incubation (TI), or PBMC treated with the TI protocol where plate passage was omitted, platelets were depleted using the a-CD41 bead kit prior to plate passage, or both of the above were performed. E, FACS analysis of phagocytic potential of CD11c+ cells within human PBMC that have either been freshly isolated (control) or TI-treated (TI) prior to overnight coincubation with PKH-labeled, 8-MOP/UVA-exposed YuCOT human melanoma cells. Bars represent average % PKH+ cells of CD11c+ cells, measured in three independent experiments. Error bars, SEM. A–C, Data summarize six independent experiments with three blood donors. D, Data summarize three independent experiments with five blood donors. A–D, Bars represent mean values, whereas error bars represent SEM. P values for each comparison calculated using paired t test; *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, differences not significant.
TI protocol with TI chamber or the clinical ECP plate rapidly induces DC maturation and unique activation profile in human PBMC, dependent on plate passage and platelets. A and B, FACS analysis of MFI change of the indicated markers from corresponding IgG controls in live CD11c+ cells among either freshly isolated PBMC (control), PBMC treated with the TI protocol and analyzed following overnight incubation (TI), or PBMC treated with the TI protocol adapted to the clinical ECP plate and analyzed following overnight incubation (ECP). For CD80, where only a subset of cells express the marker, difference in percent CD80+ cells of live CD11c+ PBMC is instead represented. C, FACS analysis of percent change of the indicated markers from corresponding IgG controls in fixed and permeabilized CD11c+ cells among either freshly isolated PBMC (control), PBMC treated with the TI protocol and analyzed following overnight incubation (TI), or PBMC treated with the TI protocol adapted to the clinical ECP plate and analyzed following overnight incubation (ECP). D, FACS analysis of TI-specific marker expression in live CD11c+ cells among either freshly isolated PBMC (control), PBMC treated with the TI protocol and analyzed following overnight incubation (TI), or PBMC treated with the TI protocol where plate passage was omitted, platelets were depleted using the a-CD41 bead kit prior to plate passage, or both of the above were performed. E, FACS analysis of phagocytic potential of CD11c+ cells within human PBMC that have either been freshly isolated (control) or TI-treated (TI) prior to overnight coincubation with PKH-labeled, 8-MOP/UVA-exposed YuCOT human melanoma cells. Bars represent average % PKH+ cells of CD11c+ cells, measured in three independent experiments. Error bars, SEM. A–C, Data summarize six independent experiments with three blood donors. D, Data summarize three independent experiments with five blood donors. A–D, Bars represent mean values, whereas error bars represent SEM. P values for each comparison calculated using paired t test; *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, differences not significant.
Although a future study using matched PBMC and tumor cells from cancer patients would be very informative, this initial investigation nevertheless allowed us to evaluate chamber-induced monocyte-to-DC conversion in a clinically relevant setting. These data strongly suggest that TI is capable of rapidly and robustly activating human monocytes into DC, with physiologic responses similar to those associated with tumor immunity in our murine models. The data also support bioequivalence between the TI chamber and the clinical ECP plate during TI, with no significant differences found in DC activation markers between the two devices.
Discussion
This study identifies the key cellular contributors to, and the nature of their collaboration in, ECP's initiation of antitumor immunity. Those insights were enabled by development and application of the first miniaturized ECP device tailored to reproduce in mice the induced clinically relevant effect in CTCL. A murine model of melanoma was chosen, to examine whether ECP's underlying immunotherapeutic principles can be potentially applied to a broader range of immunogenic cancers. After determining that established melanoma was responsive to experimental ECP, we were able to individually remove candidate cellular components to assess their contributory roles to the immunotherapeutic response.
Placed in the context of prior reports delineating ECP's platelet triggering of monocyte-to-DC maturation (8, 9), our findings reveal ECP's physiologic induction of accessible cross-presenting DC as the mechanistic centerpiece of ECP's immunizing capacity. When immunogenically loaded with apoptotic malignant cells, these new DC effectively presented tumor-derived antigens in responding recipient mice, whose CD4 and CD8 T cells collaborated to produce the defensive immune antimelanoma response. The tumor specificity of that response was demonstrated by the absence of cross-reactivity with control colorectal cancer. In short, in a therapeutic system facilitating open-ended further exploration, there is now sufficient traction to intelligently focus ECP's antitumor impact on new cancer targets, enhance its therapeutic impact, and further elucidate underlying scientific principles.
Identification of the basis of the efficacy of the roughly 6% of cancer treatments discovered through serendipity presents tantalizing scientific opportunities and daunting challenges. Deciphering therapies that, like ECP, involve complex cellular interactions can be more difficult than elucidating ones presenting as surprise responses to well-characterized pharmacologic agents (23, 24). Poststem cell allograft graft-versus-leukemia exemplifies that point (25), since it was clinically recognized by Mathé and colleagues more than 40 years ago and continues to be clarified (26). ECP, although the first FDA-approved cellular immunotherapy for any cancer (1987) and now regularly performed in more than 350 university centers worldwide, is another prime example of the challenge in resolving these cellular immunologic mechanistic mysteries (2). Introduced as a tumor-reductive method, ECP was quickly recognized to be a potent initiator of selective anticancer immunity, because its return of less than 5% of the body burden of apoptotic CTCL cells led to persistent CD8 T-cell–dependent clinical responses, accompanied by depletion of the malignant clone (3, 7), without increasing susceptibility to opportunistic infections. Yet, despite ECP's accrued clinical experience in over 60,000 patients, its mechanism proved elusive until this report. The elucidations described herein were, therefore, long awaited.
Proof of the immunotherapeutic mechanism by which ECP selectively targets patient-specific malignant CTCL cells required three advances. First, a device, scalable from mouse to man, needed to be developed. We have herein demonstrated that passage through that device, the TI chamber, affects human blood monocytes, the precursors of the required antigen-presenting cells, in the same manner as does the clinical ECP plate. Second, to determine whether ECP's antitumor impact can be extended beyond T-cell lymphoma to solid tumors, we adapted the system to the study of melanoma and colorectal cancer. Finally, to decipher the mechanism of a complex, empirically developed therapy, we need to analyze it from the result to the causes. Having demonstrated a consistent immunotherapeutic effect sufficiently potent to target established melanoma, we then individually subtracted candidate contributors to that effect, to determine their relative importance.
We have previously reported, in studies using human cells, that ECP-processed platelets adhere to the γ-chain of ECP plate–adherent fibrinogen, in a manner identical to this interaction in wound healing and blood clotting (9). These activated platelets instantaneously transpose membrane P-selectin to their surface, allowing transient monocyte docking to immobilized activated platelets and providing a platform for signaling monocytes to enter the DC maturational pathway. Our current report confirms that removal of platelets from the experimental ECP flow system reduces DC activation in human and precludes in vivo antimelanoma immunoprotection in mouse. Platelet influence thus appears to be critically necessary to physiologic induction of DC by ECP. The platelet role in the physiologic initiation of tumor-immunizing DC is a large subject in its own right and merits further detailed study, which we are actively pursuing.
The miniaturized experimental ECP system used in our experiments reproduces the effects of the conventional clinical ECP plate on processed human monocytes, lymphocytes, and tumor cells, while introducing several advantages over the current clinical device. It incorporates considerably greater treatment flexibility, by permitting individual modification of each contributing variable: flow rates through the device (controlling level of monocyte activation); chamber thickness (controlling monocyte–platelet interaction); platelet density (controlling cross-talk with monocytes); number of processed monocytes (controlling output of new DC); level of exposure of cancer cells to photoactivated 8-MOP (controlling number of apoptogenic DNA cross-links); and sparing plate-passed monocytes from exposure to photoactivated 8-MOP (polarizing the induced DC to immunizing mode). Of special importance, the insertion of a phase during which incipiently apoptotic tumor cells are coincubated with newly induced immature DC improves DC presentation of tumor-derived antigens and allows for greater control over this important step. As has been demonstrated previously, the coincubation step can significantly affect clinical anticancer efficacy of ECP in CTCL (19).
Recent advances in immune checkpoint inhibitor and chimeric antigen receptor T-cell therapy have revolutionized the field of cancer therapy (27, 28). Although conceptually more conducive to patient-specific tumor immunity, DC-based advances have encountered greater difficulties in the clinical reduction to practice (29). Our findings, superimposed on ECP's rich history in the management of advanced CTCL, suggest that the challenges of human DC therapy may relate to how physiologically DC are produced, rather than to intrinsic limitations of the potency of DC. Precursor blood monocytes have typically been directed into the DC maturational pathway through coincubation with supraphysiologic concentrations of growth stimulators and maturational cytokines, at levels far exceeding those that can be encountered by tumor-loaded DC after their reintroduction into patients (30). ECP-induced DC are more physiologically induced, without need for addition of cytokines, likely explaining their in vivo potency.
The melanoma and colorectal cancer responses to model ECP encourage enhancements of this already quite widely administered treatment. Perhaps the most immediately pressing will be testing of a revised ECP for treatment of a broader spectrum of immunogenic cancers. Because the miniaturized ECP system introduced in this article can be scaled from mouse to man, it could potentially be applicable to human therapy, permitting more flexible clinical use than the currently active device.
Disclosure of Potential Conflicts of Interest
M. Girardi has ownership interest (including patents) in Transimmune AG and is a consultant/advisory board member for Mallinckrodt Pharmaceuticals. O. Sobolev is VP Immunology at Transimmune AG. M.W. Bosenberg has expert testimony in Eli Lilly and Company. R.L. Edelson has ownership interest (including patents) in, and is a consultant/advisory board member of, Transimmune, AG. No potential conflicts of interest were disclosed by the other authors.
Authors' Contributions
Conception and design: A. Ventura, A. Vassall, E. Robinson, D. Hanlon, H. Ezaldein, M. Girardi, R.L. Edelson
Development of methodology: A. Ventura, A. Vassall, E. Robinson, R. Filler, D. Hanlon, H. Ezaldein, M.W. Bosenberg, R.L. Edelson
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): A. Ventura, A. Vassall, E. Robinson, R. Filler, K. Meeth, M.W. Bosenberg, R.L. Edelson
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): A. Ventura, A. Vassall, E. Robinson, R. Filler, D. Hanlon, H. Ezaldein, O. Sobolev, M.W. Bosenberg, R.L. Edelson
Writing, review, and/or revision of the manuscript: A. Ventura, A. Vassall, E. Robinson, R. Filler, D. Hanlon, M. Girardi, O. Sobolev, R.L. Edelson
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): A. Ventura, K. Meeth, O. Sobolev, R.L. Edelson
Study supervision: A. Ventura, D. Hanlon, R.L. Edelson
Acknowledgments
This work was supported by NIH-NCI Spore Grant 1 P50 CA121974 (R.L. Edelson and M. Girardi); NIH Cancer Center Support Grant 3 P30 CA16359-28S1 (R.L. Edelson and M. Girardi); the Howard Hughes Medical Institute training fellowship (A. Vassall); and the NY Cardiac Foundation (R.L. Edelson, A. Ventura, A. Vassall, and H. Ezaldein). Partial support was provided by R01 CA196660-01 to M.W. Bosenberg.
The authors are grateful to Dr. Robert Tigelaar for his mentoring, guidance, and experimental insight. Nicholas Theodosakis kindly helped with initial stages of the YUMM experiments. We thank our volunteer blood donors and Inger Christensen and her professional staff at Yale ECP Treatment Center for help with volunteer blood procurement. We thank our colleagues at Fraunhofer IBMT, especially Dr. Thorsten Knoll, for developing and providing the ECP-equivalent TI chamber. For technical assistance on the project, we thank Paulomi Bole Aldo for Luminex analysis, Dr. Kim Blenman and Histopathology Core for histologic analysis, Dr. Julia Lewis for FACS protocol advice, and E. Menet, G. Tokmoulina, and C. Cote at Yale FACS Core.
Special thanks is given to Sergio Chimenti, our dear friend, colleague, and mentor, for his human and professional guidance and support. His broad contributions to the Dermatology field are best measured by the lives he so very much enriched, running the full gamut from his grateful patients, to his innumerable protégés, to his dearest of friends. It is extreme privilege to be counted among latter, and we are forever deeply honored and enormously bettered by the priceless gift of our closeness to him.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.