Hepatology
Abstract
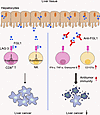
Fibrinogen-like protein 1 (FGL1) contributes to the proliferation and metabolism of hepatocytes; however, as a major ligand of the immune checkpoint, its role in the liver regional immune microenvironment is poorly understood. Hepatocytes specifically and highly expressed FGL1 under normal physiological conditions. Increases in hepatic CD8+ T and NK cell numbers and functions were found in Fgl1-deficient (Fgl1–/–) mice, but not in the spleen or lymph node, similar to findings in anti-FGL1 mAb–treated wild-type mice. Furthermore, Fgl1 deficiency or anti-FGL1 mAb blockade restrained liver metastasis and slowed the growth of orthotopic tumors, with significantly prolonged survival of tumor-bearing mice. Tumor-infiltrating hepatic CD8+ T and NK cells upregulated the expression of lymphocyte activation gene-3 (LAG-3) and exhibited stronger antitumor activities after anti-FGL1 treatment. The antitumor efficacy of FGL1 blockade depended on cytotoxic T lymphocytes and NK cells, demonstrated by using a cell-deficient mouse model and cell transfer in vivo. In vitro, FGL1 directly inhibited hepatic T and NK cells related to the receptor LAG-3. In conclusion, hepatocyte-derived FGL1 played critical immunoregulatory roles in the liver and contributed to liver metastasis and tumor growth by inhibiting CD8+ T and NK cell functions via the receptor LAG-3, providing a new strategy for liver cancer immunotherapy.
Authors
Fengjia Xi, Haoyu Sun, Hui Peng, Zhexiong Lian, Haiming Wei, Zhigang Tian, Rui Sun, Yongyan Chen

Abstract
Background Glycogen storage disease type IV (GSD IV) is an ultrarare autosomal recessive disorder that causes deficiency of functional glycogen branching enzyme and formation of abnormally structured glycogen termed polyglucosan. GSD IV has traditionally been categorized based on primary hepatic or neuromuscular involvement, with hepatic GSD IV subclassified as discrete subtypes: classic (progressive) and nonprogressive.Methods To better understand the progression of liver disease in GSD IV, we present clinical and histopathology data from 23 patients from around the world and characterized the liver involvement in the Gbe1ys/ys knockin mouse model.Results We propose an alternative to the established subtype-based terminology for characterizing liver disease in GSD IV and recognize 3 tiers of disease severity: (i) “severe progressive” liver disease, (ii) “intermediate progressive” liver disease, and (iii) “attenuated” liver disease. Analysis of liver pathology revealed that risk for liver failure cannot be predicted from liver biopsy findings alone in individuals affected by GSD IV. Moreover, analysis of postmortem liver pathology from an individual who died over 40 years after being diagnosed with nonprogressive hepatic GSD IV in childhood verified that liver fibrosis did not regress. Last, characterization of the liver involvement in a mouse model known to recapitulate the adult-onset neurodegenerative form of GSD IV (Gbe1ys/ys mouse model) demonstrated hepatic disease.Conclusion Our findings challenge the established subtype-based view of GSD IV and suggest that liver disease severity among patients with GSD IV represents a disease continuum.Trial registration ClinicalTrials.gov NCT02683512Funding None
Authors
Rebecca L. Koch, Bridget T. Kiely, Su Jin Choi, William R. Jeck, Leticia S. Flores, Vikrant Sood, Seema Alam, Gilda Porta, Katy LaVecchio, Claudia Soler-Alfonso, Priya S. Kishnani
Abstract
Portal hypertension (PHTN) is a severe complication of liver cirrhosis and is associated with intrahepatic sinusoidal remodeling induced by sinusoidal resistance and angiogenesis. Collagen type IV (COL4), a major component of basement membrane, forms in liver sinusoids upon chronic liver injury. However, the role, the cellular source and expression regulation of COL4 in liver diseases is unknown. Here, we examined how COL4 is produced and how it regulates sinusoidal remodeling in fibrosis and PHTN. Human cirrhotic liver sample RNA-sequencing showed increased COL4 expression, which was further confirmed via immunofluorescence staining. scRNA-sequencing identified liver sinusoidal endothelial cells (LSECs) as the predominant source of COL4 upregulation in mouse fibrotic liver. In addition, COL4 was upregulated in a tumor necrosis factor α–nuclear factor–κB dependent manner through an epigenetic mechanism in liver sinusoidal endothelial cells in vitro. Indeed, by utilizing a CRISPRi-dCas9-KRAB-mediated epigenome editing approach, epigenetic repression of the enhancer-promoter interaction showed silencing of COL4 gene expression. LSEC-specific COL4 gene mutation or repression in vivo abrogated sinusoidal resistance and angiogenesis, which thereby alleviated sinusoidal remodeling and PHTN. Our findings reveal that LSECs promote sinusoidal remodeling and PHTN during liver fibrosis through COL4 deposition.
Authors
Can Gan, Usman Yaqoob, Jianwen Lu, Man Xie, Abid A. Anwar, Nidhi Jalan-Sakrikar, Sofia Jerez, Tejasav Sehrawat, Amaia Navarro-Corcuera, Enis Kostallari, Nawras W. Habash, Sheng Cao, Vijay H. Shah
Abstract
Manganese is an essential yet potentially toxic metal. Initially reported in 2012, mutations in SLC30A10 are the first known inherited cause of manganese excess. SLC30A10 is an apical membrane protein that exports manganese from hepatocytes into bile and from enterocytes into the lumen of the gastrointestinal tract. SLC30A10 deficiency results in impaired gastrointestinal manganese excretion, leading to manganese excess, neurologic deficits, liver cirrhosis, polycythemia, and erythropoietin excess. Neurologic and liver disease are attributed to manganese toxicity. Polycythemia is attributed to erythropoietin excess. The goal of this study was to determine the basis of erythropoietin excess in SLC30A10 deficiency. Here we demonstrate that transcription factors hypoxia-inducible factor 1a (Hif1a) and 2a (Hif2a), key mediators of the cellular response to hypoxia, are both upregulated in livers of Slc30a10-deficient mice. Hepatic Hif2a deficiency corrected erythropoietin expression and polycythemia and attenuated aberrant hepatic gene expression in Slc30a10-deficient mice, while hepatic Hif1a deficiency had no discernible impact. Hepatic Hif2a deficiency also attenuated manganese excess, although the underlying cause of this is not clear at this time. Overall, our results indicate that hepatic HIF2 is a key determinant of pathophysiology in SLC30A10 deficiency and expand our understanding of the contribution of HIFs to human disease.
Authors
Milankumar Prajapati, Jared Z. Zhang, Lauren Chiu, Grace S. Chong, Courtney J. Mercadante, Heather L. Kowalski, Bradley S. Delaney, Jessica A Anderson, Shuling Guo, Mariam Aghajan, Thomas B. Bartnikas
Abstract
Diagnostic challenges continue to impede development of effective therapies for successful management of alcohol-associated hepatitis (AH), thus creating an unmet need to identify and develop non-invasive biomarkers for AH. In murine models of ethanol-induced liver injury, complement activation contributes to hepatic inflammation and injury. Therefore, we hypothesized that complement proteins could be rational diagnostic/prognostic biomarkers in AH. Here, we performed a comparative analysis of data derived from the human hepatic and serum proteome to identify and characterize complement protein signatures in severe AH (sAH). The quantity of multiple complement proteins was perturbed in liver and serum proteome of patients with sAH. Multiple complement proteins differentiated patients with sAH from those with alcohol cirrhosis (AC), alcohol use disorder (AUD) and healthy controls (HCs). Notably, serum collectin 11 and C1q binding protein were strongly associated with sAH and exhibited good discriminatory performance amongst patients with sAH, AC, AUD, and HCs. Furthermore, complement component receptor 1-like protein (CR1L) was negatively associated with pro-inflammatory cytokines. Additionally, lower serum mannose-binding lectin associated serine protease 1 and coagulation factor II were associated with and independently predicted 90-day mortality. In summary, meta-analysis of proteomic profiles from liver and circulation revealed complement protein signatures of sAH, highlighting a complex perturbation of complement and identifying potential diagnostic and prognostic biomarkers for patients with sAH.
Authors
Moyinoluwa T. Taiwo, Emily Huang, Vai Pathak, Annette Bellar, Nicole Welch, Jaividhya Dasarathy, David Streem, Craig J. McClain, Mack C. Mitchell, Bruce A. Barton, Gyongyi Szabo, Srinivasan Dasarathy, Esperance A. Schaefer, Jay Luther, Le Z. Day, Xinshou Ouyang, Suyavaran Arumugam, Wajahat Z. Mehal, Jon M. Jacobs, Russell P. Goodman, Daniel M. Rotroff, Laura E. Nagy
Abstract
Excessive lipolysis in white adipose tissues (WAT) leads to insulin resistance (IR) and ectopic fat accumulation in insulin-sensitive tissues. However, the impact of Gi-coupled receptors in restraining adipocyte lipolysis through inhibition of cAMP production remained poorly elucidated. Given that the Gi-coupled P2Y13 receptor (P2Y13-R) is a purinergic receptor expressed in WAT, we investigated its role in adipocyte lipolysis and its effect on IR and metabolic dysfunction-associated steatotic liver disease (MASLD). In human, mRNA expression of P2Y13-R in WAT was negatively correlated to adipocytes lipolysis. In mice, adipocytes lacking P2Y13-R displayed higher intracellular cAMP levels, indicating impaired Gi signaling. Consistently, the absence of P2Y13-R was linked to increased lipolysis in adipocytes and WAT explants via hormone-sensitive lipase activation. Metabolic studies indicate that mice lacking P2Y13-R show a greater susceptibility to diet-induced IR, systemic inflammation, and MASLD compared to their wild-type counterparts. Assays conducted on precision-cut liver slices exposed to WAT conditioned medium and on liver-specific P2Y13-R knockdown mice suggested that P2Y13-R activity in WAT protects from hepatic steatosis, independently of liver P2Y13-R expression. In conclusion, our findings support the idea that targeting adipose P2Y13-R activity may represent a pharmacological strategy to prevent obesity-associated disorders, including type 2 diabetes and MASLD.
Authors
Thibaut Duparc, Emilia Gore, Guillaume Combes, Diane Beuzelin, Julie Pires Da Silva, Vanessa Bouguetoch, Marie-Adeline Marquès, Ana Velazquez, Nathalie Viguerie, Geneviève Tavernier, Peter Arner, Mikael Rydén, Dominique Langin, Nabil Sioufi, Mohamad Nasser, Cendrine Cabou, Souad Najib, Laurent O. Martinez
Abstract
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene lead to cystic fibrosis (CF), a life-threating autosomal recessive genetic disease. While recently approved Trikafta dramatically ameliorates CF lung diseases, there is still a lack of effective medicine to treat CF-associated liver disease (CFLD). To address this medical need, we used a recently established CF rabbit model to test if Sotagliflozin, a Sodium-Glucose cotransporter 1 and 2 (SGLT1/2) inhibitor drug that is approved to treat diabetes, can be repurposed to treat CFLD. Sotagliflozin treatment led to systemic benefits to CF rabbits, evidenced by increased appetite and weight gain as well as prolonged lifespan. For CF liver related phenotypes, the animals benefited from normalized blood chemistry and bile acid parameters. Further, Sotagliflozin alleviated non-alcoholic steatohepatitis (NASH)-like phenotypes including liver fibrosis. Intriguingly, Sotagliflozin treatment markedly reduced the otherwise elevated endoplasmic reticulum (ER) stress responses in the liver and other affected organs of CF rabbits. In summary, our work demonstrates that Sotagliflozin attenuates liver disorders in CF rabbits, and merits Sotagliflozin as a potential drug to treat CFLD.
Authors
Xiubin Liang, Xia Hou, Mohamad Bouhamdan, Yifei Sun, Zhenfeng Song, Carthic Rajagopalan, Hong Jiang, Hong-Guang Wei, Jun Song, Dongshan Yang, Yanhong Guo, Yihan Zhang, Hongmei Mou, Jifeng Zhang, Y. Eugene Chen, Fei Sun, Jian-Ping Jin, Kezhong Zhang, Jie Xu
Abstract
Depletion of torsinA from hepatocytes leads to reduced liver triglyceride secretion and marked hepatic steatosis. TorsinA is an atypical ATPase that lacks intrinsic activity unless it is bound to its activators lamina-associated polypeptide 1 (LAP1) or luminal domain-like LAP1 (LULL1). We previously demonstrated that depletion of LAP1 from hepatocytes has more modest effects on liver triglyceride secretion and steatosis development than depletion of torsinA. We now show that depletion of LULL1 alone does not significantly decrease triglyceride secretion or cause steatosis. However, simultaneous depletion of both LAP1 and LULL1 leads to defective triglyceride secretion and marked steatosis similar to that observed with depletion of torsinA. Depletion of both LAP1 and torsinA from hepatocytes generated phenotypes similar to those observed with only torsinA depletion, implying that the two proteins act in the same pathway in liver lipid metabolism. Our results demonstrate that torsinA and its activators dynamically regulate hepatic lipid metabolism.
Authors
Antonio Hernandez-Ono, Yi Peng Zhao, John W. Murray, Cecilia Östlund, Michael J. Lee, Angsi Shi, William T. Dauer, Howard J. Worman, Henry N. Ginsberg, Ji-Yeon Shin
Abstract
Intrahepatic macrophages in nonalcoholic steatohepatitis (NASH) are heterogenous and include proinflammatory recruited monocyte derived macrophages. The receptor for advanced glycation end products (RAGE) is expressed on macrophages and can be activated by damage associated molecular patterns (DAMPs) upregulated in NASH, yet the role of macrophage-specific RAGE signaling in NASH is unclear. Therefore, we hypothesized that RAGE expressing macrophages are proinflammatory and mediate liver inflammation in NASH. Compared to healthy controls, RAGE expression was increased in liver biopsies from human NASH patients. In a high -fat, -fructose, and -cholesterol (FFC)-induced murine model of NASH, RAGE expression was increased, specifically on recruited macrophages. FFC mice that received a pharmacological inhibitor of RAGE (TTP488), and myeloid-specific RAGE knockout mice (RAGE-MKO) had attenuated liver injury associated with a reduced accumulation of RAGE+ recruited macrophages. Transcriptomic analysis suggested that pathways of macrophage and T-cell activation were upregulated by FFC diet, inhibited by TTP488 treatment, and reduced in RAGE-MKO mice. Correspondingly, the secretome of ligand-stimulated bone marrow derived macrophages from RAGE-MKO mice had an attenuated capacity to activate CD8+ T cells. Our data implicate RAGE as what we propose to be a novel and potentially targetable mediator of the proinflammatory signaling of recruited macrophages in NASH.
Authors
Gopanandan Parthasarathy, Amy S. Mauer, Naresh Golla, P. Vineeth Daniel, Lily H. Kim, Guneet S. Sidhu, George W. Marek 3rd, Emilien Loeuillard, Anuradha Krishnan, Hyun Se Kim Lee, Kevin D. Pavelko, Michael Charlton, Petra Hirsova, Sumera I. Ilyas, Harmeet Malhi
Abstract
Drug-induced liver injury (DILI), especially acetaminophen overdose, is the leading cause of acute liver failure. Pregnane X receptor (PXR) is a nuclear receptor and the master regulator of drug metabolism. Aberrant activation of PXR plays a pathogenic role in the acetaminophen hepatotoxicity. Here, we aimed to examine the PXR S-nitrosylation (SNO) in response to acetaminophen. We found that PXR was S-nitrosylated in hepatocytes and the mouse livers after exposure to acetaminophen or S-nitrosoglutathione (GSNO). Mass-spectrometry and site-directed mutagenesis identified the cysteine 307 as the primary residue for SNO-modification. In hepatocytes, SNO suppressed both agonist (rifampicin and SR12813)-induced and constitutively active PXR (VP-PXR) activations. Furthermore, in acetaminophen overdosed mouse livers, PXR protein was decreased at the centrilobular regions overlapping with increased SNO. In PXR-deficient (PXR-/-) mice, replenishing the livers with the SNO-deficient PXR significantly aggravated hepatic necrosis, increased HMGB1 release, and exacerbated liver injury and inflammation. Particularly, we demonstrated that S-nitrosoglutathione reductase (GSNOR) inhibitor N6022 promoted hepatoprotection by increasing the levels of PXR S-nitrosylation. In conclusion, PXR is post-translationally modified by S-nitrosylation in hepatocytes in response to acetaminophen. This modification mitigated the acetaminophen-induced PXR hyperactivity. It may serve as a target for therapeutical intervention.
Authors
Qi Cui, Tingting Jiang, Xinya Xie, Haodong Wang, Lei Qian, Yanyan Cheng, Qiang Li, Tingxu Lu, Qinyu Yao, Jia Liu, Baochang Lai, Chang Chen, Lei Xiao, Nanping Wang
No posts were found with this tag.


